Citation Information
La Vecchia, JoLS-Pub J Life Sci. Vol.3, No.1, January 2026:7-12 [https://doi.org/qnxc]
Acute myeloid leukemia (AML) is a rapidly progressing cancer characterized by the uncontrolled growth of immature myeloid cells, precursor cells in the bone marrow, which crowd out normal blood cells and impair their production. It remains one of the deadliest forms of adult leukemia and is the second most common among children. This poor prognosis is largely due to AML’s ability to develop resistance to frontline chemotherapy drugs.
One of the most promising recent therapies is Venetoclax, a drug approved by the US Food and Drug Administration (FDA) that targets the BCL-2 protein to trigger cancer cell death (1). It is a BH3 mimetic drug that forces tumor cells to self-destruct by blocking their survival proteins. Venetoclax is often used alone or in combination with other treatments, such as the hypomethylating agent Azacitidine (2-4). Unfortunately, many patients relapse because leukemia cells adapt to survive by exploiting the dynamic nature of their mitochondria. Although advances in treatment have improved outcomes, survival rates remain limited, with only about 30% of adults and 65% of children surviving at least five years after diagnosis, highlighting the ongoing challenge (5).
Cells adapt through various mechanisms: how do these cells become resistant?
They do so with the help of small organelles called mitochondria. These organelles do much more than act as the “powerhouse of the cell”; they control stress responses and determine whether the cell survives or undergoes programmed cell death (6-8).
Our recent studies show that resistance to BH3 mimetic drugs, like Venetoclax, is strongly linked to mitochondrial morphology, amount, and organelle interactions (9, 10). Central to this mitochondrial adaptation is a protein called OPA1, the master regulator of cristae architecture, which shapes the folds inside mitochondria known as cristae (11, 12). In therapy-resistant AML cells, OPA1 expression is increased, leading to tighter and more numerous mitochondrial folds. This remodeling traps critical molecules like cytochrome c inside the mitochondria, preventing the usual chain reaction that causes programmed cell death, or apoptosis (13). This research focuses on whether small molecules that inhibit OPA1 (14) can reverse these mitochondrial changes to overcome Venetoclax resistance in leukemia. Mechanistically, the study also explores the metabolic flexibility hidden behind the loss of OPA
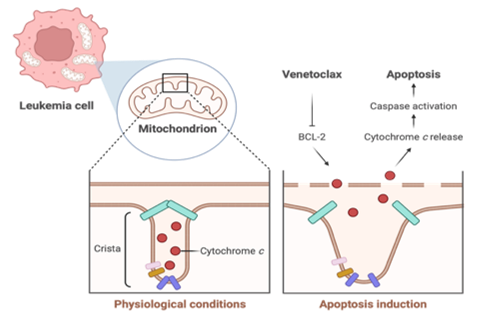
Fig. 1. Mitochondrial remodeling drives apoptosis in leukemia cells.
Schematic representation of mitochondrial cristae structure and cytochrome c release during apoptosis induction in leukemia cells. Under physiological conditions (left panel), cytochrome c is within the cristae of the inner mitochondrial membrane. Apoptosis induction with Venetoclax (right panel) inhibits BCL-2, leading to cristae remodeling and cytochrome c release into the cytosol, which activates caspases and promotes programmed cell death (12, 15).
How can we effectively target mitochondrial dynamics, specifically OPA1-mediated remodeling, to overcome drug resistance in leukemia?
Mitochondria as a target!
Researchers combined cutting-edge CRISPR screening, a gene-editing technique that systematically knocks out thousands of genes in leukemia cells to identify which ones are essential for survival, with electron microscopy. Together, they unravel how leukemia cells escape treatment, pinpointing OPA1 as a key factor for this survival advantage. Electron microscopy provided a close-up look at the mitochondrial inner membrane architecture in AML cells, highlighting cristae structures central to OPA1’s function. OPA1 reshapes the inner mitochondrial membrane to sequester apoptotic triggers, effectively shielding leukemia cells from chemotherapeutic attack. This remodeling traps cytochrome c, a molecule pivotal for triggering apoptosis - the programmed cell death chemotherapy aims to induce, even after Venetoclax treatment.
Patient-derived AML samples (PDXs) and cell models revealed that therapy-resistant cells had unusually tight and numerous cristae, a condition accompanying elevated OPA1 protein levels.
Inhibiting OPA1 with two experimental small-molecule drugs, MYLS22 and its more potent derivative Opitor-0, successfully disrupted these mitochondrial folds (Fig. 2). These inhibitors have been tested in vitro (in cell models) and in mouse models of different solid cancers (16-19). The prototypical OPA1 inhibitor MYLS22 has also been used as monotherapy in the context of AML, where it causes cell cycle arrest through regulating reactive oxygen species (ROS) production (20).
To evaluate the impact on healthy mitochondria, we compared mitochondrial integrity in untreated versus MYLS22-treated hematopoietic cells from mice. MYLS22 exhibited a favorable safety profile in the mouse hematopoietic system, with no systemic adverse effects observed in CD34+ cells.
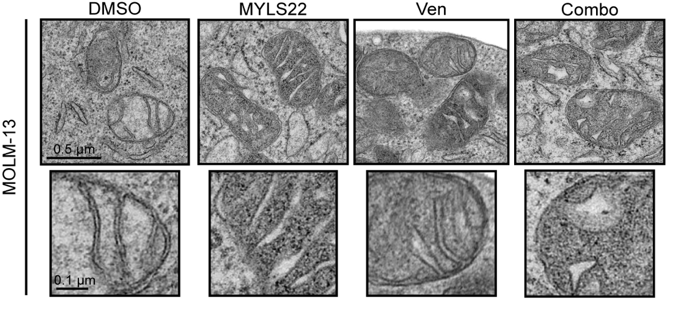
Fig. 2. Pharmacological OPA1 inhibition impairs cristae morphology and sensitizes human AML to apoptosis.
Representative electron micrographs of MOLM-13 treated for 6 hours with: 40 μM MYLS22, 6 nM Venetoclax, their combination (combo), or dimethyl sulfoxide (DMSO). Scale bars, 0.5 or 0.1 μm.
Cristae width is increased following OPA1 inhibition.
Connecting the dots: what our data tells us
Despite the previous data indicating the OPA1 inhibitors, MYLS22 and Opitor-0 may have an anti-cancer effect, the mechanism of action by which they act and the potential of these compounds for the treatment of AML remains unclear.
This disruption of mitochondrial structure by OPA1 inhibitors allowed cytochrome c to escape, significantly amplifying the cell death effects of Venetoclax. These initial results built a compelling case that OPA1-driven cristae changes are central to why some leukemia cells resist therapy.
In several AML PDX models and patient samples, combining OPA1 inhibitors with Venetoclax showed more than doubled survival times compared to the single treatment, across diverse AML subtypes, independent of the mutational background. Clinically relevant, MYLS22 and Opitor-0 efficiently restore sensitivity to Venetoclax even in apoptosis-resistant AML, harboring TP53 mutations. TP53 is the "guardian of the genome" gene that normally stops leukemia cells from growing out of control by fixing DNA damage or triggering their self-destruction, but when mutated in AML, it lets cancer cells survive treatments and spread aggressively. TP53 mutations in AML are associated with the shortest overall survival (21, 22) and strongly correlate with Venetoclax resistance (9, 10, 23), complicating the treatment of this subgroup of patients.
Since AML is a heterogeneous disease involving countless mutations, combinatorial therapeutic approaches improve treatment outcomes not only for patients with leukemia but also for individuals with other cancer types.
Beyond its role in cristae architecture and apoptosis regulation, the precise role of OPA1 in AML biology was unknown. Loss of OPA1 function forced leukemia cells to rely heavily on glutamine metabolism for survival and made them more susceptible to ferroptosis, an iron-dependent form of cell death involving damaging lipid oxidation. This dual mechanism suggests that OPA1 inhibitors not only control mitochondrial shape but also trigger metabolic stress that cancer cells cannot easily overcome. Importantly, these inhibitors did not impair normal blood cell development in preclinical safety tests, underscoring their potential as safe, targeted therapies.
Together, these findings identify OPA1 as a promising therapeutic target to overcome drug resistance in AML and possibly other cancers characterized by abnormal mitochondrial dynamics. Importantly, mitochondrial remodeling and OPA1 upregulation are observed in various tumors, suggesting these therapeutic insights may apply broadly, heralding a new class of mitochondria-targeting drugs.
This study presents a novel strategy to address chemoresistance in AML by modulating the mitochondrial dynamics regulator OPA1. Combining OPA1 inhibitors with Venetoclax enhanced leukemia cell elimination and markedly improved survival in preclinical models. These promising findings highlight the clinical potential of refining OPA1-targeted therapies and support their continued development as part of next-generation treatments for refractory AML and other cancers with altered mitochondrial function.
What's Next? Let’s bring our drugs from bench to bedside
The discovery and validation of the OPA1 inhibitors MYLS22 and Opitor-0 mark a significant advance in overcoming drug resistance in acute myeloid leukemia. These compounds, developed and optimized in the laboratory, have shown that by directly remodeling mitochondrial structure, they can restore sensitivity to Venetoclax and reliably trigger programmed cell death, even in models of AML that are notoriously resistant to therapy. Future work will focus on refining Opitor-0 as the more potent compound, conducting comprehensive toxicology studies, and initiating early-phase clinical trials. Preclinical studies will also expand to cover a wider range of leukemia subtypes and mutational backgrounds, with particular focus on “difficult to treat” groups such as those with driver mutations like TP53.
Researchers will continue to explore metabolic vulnerabilities induced by OPA1 inhibition, including glutamine dependency, reliance on glutamine for fuel, and ferroptosis susceptibility, sensitivity to iron-triggered cell death, which could enable new combination therapies for improved patient outcomes.
Together, these efforts lay the foundation for clinical trials that could introduce a new class of mitochondria-targeting therapeutics, potentially transforming treatment for AML and other aggressive cancers by exploiting mitochondrial remodeling as an Achilles’ heel (Fig. 3).
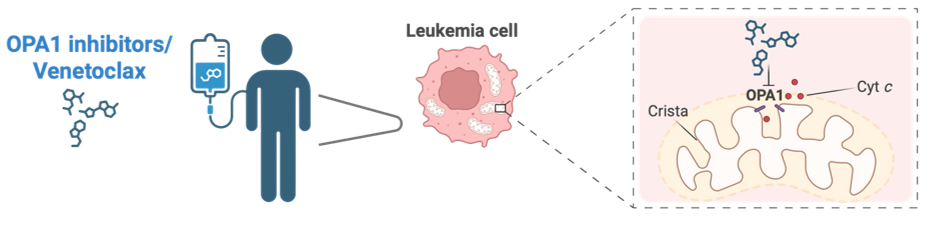
Fig. 3. Combination of OPA1 inhibitors and Venetoclax disrupts mitochondrial structure to overcome therapy resistance in leukemia cells.
Schematic representation of OPA1 inhibitors/venetoclax to the patient, their action on leukemia cells, and the molecular mechanism at the mitochondrial level.
Combination therapy with OPA1 inhibitors and Venetoclax targets leukemia cells by disrupting mitochondrial cristae structure, promoting cytochrome c release, and inducing cell death.
Notes: Parts of the figures in this article were created with BioRender.com.
References
1. L. Campos, High expression of bcl-2 protein in acute myeloid leukemia cells is associated with poor response to chemotherapy. Blood 81, 3091-3096 (1993).
2. C. D. DiNardo, Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood 133, 7-17 (2019).
3. C. D. DiNardo, Molecular patterns of response and treatment failure after frontline venetoclax combinations in older patients with AML. Blood 135, 791-803 (2020).
4. D. A. Pollyea, Venetoclax for AML: changing the treatment paradigm. Blood Adv 3, 4326-4335 (2019).
5. R. L. Siegel, Cancer statistics, 2024. CA Cancer J Clin 74, 12-49 (2024).
6. G. W. Dorn, Evolving Concepts of Mitochondrial Dynamics. Annu Rev Physiol 81, 1-17 (2019).
7. M. Giacomello, The cell biology of mitochondrial membrane dynamics. Nat Rev Mol Cell Biol 21, 204-224 (2020).
8. S. Doshi, Mitochondrial Dynamics in Blood Cancer Development and Progression. Curr Pharmacol Rep 11, 53 (2025).
9. X. Chen, Targeting Mitochondrial Structure Sensitizes Acute Myeloid Leukemia to Venetoclax Treatment. Cancer Discov 9, 890-909 (2019).
10. C. Glytsou, Mitophagy Promotes Resistance to BH3 Mimetics in Acute Myeloid Leukemia. Cancer Discov 13, 1656-1677 (2023).
11. C. Glytsou, Optic Atrophy 1 Is Epistatic to the Core MICOS Component MIC60 in Mitochondrial Cristae Shape Control. Cell Rep 17, 3024-3034 (2016).
12. C. Frezza, OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell 126, 177-189 (2006).
13. S. La Vecchia, Small-molecule OPA1 inhibitors reverse mitochondrial adaptations to overcome therapy resistance in acute myeloid leukemia. Sci Adv 11, eadx8662 (2025).
14. A. Pellattiero, Small molecule OPA1 inhibitors amplify cytochrome c release and reverse cancer cells resistance to Bcl-2 inhibitors. Sci Adv 11, eadx4562 (2025).
15. L. Scorrano, A distinct pathway remodels mitochondrial cristae and mobilizes cytochrome c during apoptosis. Dev Cell 2, 55-67 (2002).
16. M. Noguchi, Inhibition of the mitochondria-shaping protein Opa1 restores sensitivity to Gefitinib in a lung adenocarcinomaresistant cell line. Cell Death Dis 14, 241 (2023).
17. S. Herkenne, Developmental and Tumor Angiogenesis Requires the Mitochondria-Shaping Protein Opa1. Cell Metab 31, 987-1003 e1008 (2020).
18. M. L. Baek, Mitochondrial structure and function adaptation in residual triple negative breast cancer cells surviving chemotherapy treatment. Oncogene 42, 1117-1131 (2023).
19. A. Diokmetzidou, Metastatic breast cancer cells are selectively dependent on the mitochondrial cristae-shaping protein OPA1. Cell Death Dis 16, 539 (2025).
20. C. Larrue, Mitochondrial fusion is a therapeutic vulnerability of acute myeloid leukemia. Leukemia 37, 765-775 (2023).
21. M. Chomczyk, Impact of p53-associated acute myeloid leukemia hallmarks on metabolism and the immune environment. Front Pharmacol 15, 1409210 (2024).
22. G. Qin, The Prognostic Value of TP53 Mutations in Adult Acute Myeloid Leukemia: A Meta-Analysis. Transfus Med Hemother 50, 234-244 (2023).
23. R. Pan, Synthetic Lethality of Combined Bcl-2 Inhibition and p53 Activation in AML: Mechanisms and Superior Antileukemic Efficacy. Cancer Cell 32, 748-760 e746 (2017).
Author:
| Dr. La Vecchia is a Postdoctoral scientist at Rutgers University. Her work focuses on cancer immunology, cancer metabolism and drug discovery |

Reviewers:
| Dr. Spicer is a Postdoctoral Research Fellow at Vanderbilt University Medical Center, studying the effects of systemic inflammation in persistent cognitive decline and the onset and progression of neurodegenerative disease. |
| Alec Kramer is a neuroscience PhD candidate in the Neuroscience Graduate Program at Vanderbilt University. |
Katie Shapiro | Katie Shapiro is an undergraduate student at Vanderbilt University, mentored by Dr. Fiona Harrison. |