Citation Information
Pedretti et al., JoLS-Pub J Life Sci. Vol.2, No.12, December 2025:1-5 [https://doi.org/qnxb]
Introduction :
Acknowledgements
Imagine a disease that hides in plain sight for years, ultimately causing serious conditions like heart failure and/or peripheral nerve numbness, yet is rarely caught early enough to treat effectively. This is the clinical reality of the estimated 500,000 patients living with transthyretin amyloidosis (ATTR amyloidosis).1,2 ATTR amyloidosis is a progressive and fatal condition caused by the misfolding of a protein called transthyretin. Transthyretin, or TTR, is mostly produced in the liver and is normally responsible for transporting thyroxine and retinol throughout the body. When TTR misfolds, it aggregates to form long, rod-like structures we call amyloid fibrils. These fibrils accumulate in virtually every organ in the body, particularly the heart and peripheral nerves. The consequences of this accumulation include heart failure, sensory loss, and eventual death in 5-10 years if left untreated.3 There are currently 5 FDA-approved drugs on the market for ATTR amyloidosis, but studies have shown that these treatments are most effective when administered early in the disease course.4, 5 Unfortunately, diagnosing ATTR amyloidosis is a long and difficult process lasting 3 to 7 years after symptoms first appear, and often requiring invasive biopsies or advanced imaging tests that are only available in specialized clinics.1, 3 Our research seeks to address the current need for an early ATTR amyloidosis detection tool by using a structure-guided approach to develop a simple, accurate, and non-invasive blood test capable of identifying ATTR amyloidosis in its earliest stages, when patients are still asymptomatic or beginning to experience subtle warning signs.
Research questions that we addressed :
Although significant progress has been made in understanding the biology and therapeutic targets of ATTR, critical gaps remain in our knowledge. We set out to answer the following key questions:
• Do disease-causing aggregates of TTR circulate in the bloodstream?
• Do these aggregates only appear in patients with clinically confirmed ATTR, and can they be distinguished from normal TTR?
• Are the aggregates detectable in all forms of the disease, including both hereditary (variant) and age-associated (wild-type) ATTR amyloidosis?
• Can we design a tool that binds exclusively to the toxic, misfolded conformation of TTR found in amyloid fibrils?
The objective of our research :
Our goal was to design a blood-based detection tool that identifies TTR aggregates, the early culprits of ATTR amyloidosis, using the structural information from mature patient-derived cardiac fibrils. We hypothesized that TTR aggregation begins in the blood, and that early aggregates share structural features with the mature amyloid fibrils, which we previously characterized structurally.6,7 We further hypothesized that we could use these features to design a probe that binds only the disease-causing aggregates and not the normal TTR protein.
As summarized in Figure 1, we created a small molecule-based probe that :
• Specifically recognizes TTR in its misfolded, disease-causing form, named ATTR aggregates.
• Detects even small quantities of these aggregates in patient plasma
• Works across both hereditary (ATTRv) and wild-type (ATTRwt) amyloidoses forms of the disease
• Correlates with disease status and responds to treatment
We called this tool TAD1, short for “Transthyretin Aggregation Detector 1”.
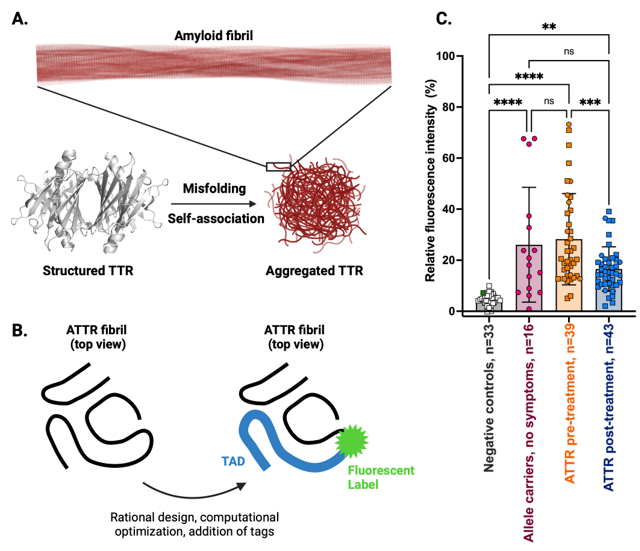
Figure 1. A. Disease mechanism underlying transthyretin amyloidosis (ATTR). In ATTR, transthyretin (TTR) loses its proper shape (misfolds) and sticks to itself, forming long, harmful fibers called amyloid fibrils. These fibrils build up in organs and cause disease symptoms ranging from heart failure to peripheral nerve damage. B. Structure-based design of Transthyretin Aggregation Detector 1 (TAD1). Using the molecular information from detailed 3D maps of patient-derived ATTR fibrils, we designed a small molecule called TAD1 that attaches only to the harmful, misfolded version of TTR. We added a fluorescent tag to the probe, which lights up when it binds to the dangerous protein clumps, allowing us to measure their concentration in a sample. C. Discovery of aggregated TTR in blood of ATTR patients. This chart shows how much TAD1 fluoresces or “lights up” in blood samples from different people. Healthy controls and patients with other diseases (negative controls) such as light chain amyloidosis show very little signal. People with genetic risk (allele carriers) but no symptoms have slightly higher signals, suggesting the use of TAD1 as a diagnostic tool to detect disease prior to symptom onset. Patients with ATTR have much higher signals before treatment, and their signals drop significantly after treatment, showing that therapy is working and that TAD1 can also be used to monitor treatment response.
Answers we found :
Our work across three studies provides several key findings:
1. TAD1 specifically binds disease-causing TTR conformations but not normal, functional TTR conformations. It is sensitive enough to detect nanogram amounts of aggregates and produces no significant signal in healthy controls or non-ATTR heart failure cases.7, 8
2. TAD1 detects aggregates in both wild-type and variant forms of ATTR, including the most prevalent TTR mutations V122I and V30M. It performs consistently regardless of the organ primarily affected (heart and/or nerves).8, 9
3. Aggregates detected by TAD1 decrease significantly after treatment, supporting its use as a tool to monitor therapeutic response.7, 8, 10
4. Notably, some asymptomatic carriers of pathogenic TTR variants exhibited TAD1-positive plasma, raising the possibility of early detection before clinical onset.8
5. Structural studies confirmed that aggregation-driving segments of TTR used to design TAD1 are structurally consistent across all patient-derived fibrils examined.6, 7 This gives TAD1 broad applicability in diverse patient populations.
Implications of our findings:
These results introduce a new way to detect ATTR, one that is based not on symptoms or genetic risk alone, but on the actual disease mechanism—protein aggregation. The implications are profound:
• We developed a non-invasive, plasma-based method to detect ATTR at early or even pre-symptomatic stages.
• This tool may enable treatment monitoring, allowing clinicians to objectively measure whether or not therapy is working.
• It has potential for large-scale screening of high-risk populations, such as older adults and carriers of common pathogenic variants.
• From a research perspective, TAD1 opens the door to studying aggregation biology in animal models and/or further patient-derived samples, accelerating biomarker and drug development.
Overall, TAD1 gives us a tool to better study a poorly understood disease and, as it is further developed into a clinical tool, help physicians diagnose and treat ATTR patients much earlier and more effectively. This structure-based approach could also ultimately serve as a model for other protein misfolding disorders, including Alzheimer’s and Parkinson’s diseases.
Limitations of our research:
Several important limitations remain:• The sample size across all different patient populations is still modest and therefore requires additional larger studies to fully account for the full diversity of ATTR amyloidosis presentations.
• There is limited longitudinal data. Further studies are needed to track aggregate levels over time within the same patient and in response to different therapies.
• We have yet to assess whether other misfolded TTR species exist in blood that TAD1 cannot detect.
Questions that remain to be addressed in future research:
Many open questions continue to drive our future studies:
• How soon prior to disease onset can we detect ATTR aggregates?
• How do patient demographics impact the levels of ATTR aggregates in plasma?
• Does the type of treatment impact plasma ATTR aggregate levels? Does reduced ATTR species correlate with patient prognosis?
• What are the biochemical/structural components of ATTR aggregates? Do these characteristics vary depending on clinical phenotype?
• What is the source of ATTR aggregates in plasma?
Acknowledgements
We are grateful to the many patients who contributed samples to this research and to our collaborators at the Indiana University, Cleveland Clinic, Oregon Health & Science University, Columbia University, Boston Medical Center, and UT Southwestern. Special thanks to the Saelices Lab and our funding sources including the American Heart Association Career Development Award, NIH Director’s New Innovator Award (1DP2HL163810-01), and The Welch Foundation.
References :
1 Hellenbart, E. L., Ipema, H. J., Rodriguez‐Ziccardi, M. C., Krishna, H. & Didomenico, R. J. Disease‐modifying therapies for amyloid transthyretin cardiomyopathy: Current and emerging medications. Pharmacotherapy: The Journal of Human Pharmacology and Drug Therapy 45, 124-144 (2025). https://doi.org:10.1002/phar.4639
2 Abouezzeddine, O. F. et al. Prevalence of Transthyretin Amyloid Cardiomyopathy in Heart Failure With Preserved Ejection Fraction. JAMA Cardiology 6, 1267 (2021). https://doi.org:10.1001/jamacardio.2021.3070
3 Ruberg, F. L. & Maurer, M. S. Cardiac Amyloidosis Due to Transthyretin Protein. JAMA 331, 778 (2024). https://doi.org:10.1001/jama.2024.0442
4 Correia-Rodrigues, Â. et al. From pathophysiology to treatment in transthyretin cardiac amyloidosis. Trends in Molecular Medicine 0 (2025). https://doi.org:10.1016/j.molmed.2025.07.002
5 Griffin, J. M. et al. ATTR Amyloidosis: Current and Emerging Management Strategies: JACC: CardioOncology State-of-the-Art Review. JACC: CardioOncology 3 (2021/10/01). https://doi.org:10.1016/j.jaccao.2021.06.006
6 Nguyen, B. A. et al. Structural polymorphism of amyloid fibrils in ATTR amyloidosis revealed by cryo-electron microscopy. Nature Communications 15 (2024). https://doi.org:10.1038/s41467-024-44820-3
7 Pedretti, R. et al. Structure-Based Probe Reveals the Presence of Large Transthyretin Aggregates in Plasma of ATTR Amyloidosis Patients. JACC: Basic to Translational Science 9, 1088-1100 (2024). https://doi.org:10.1016/j.jacbts.2024.05.013
8 Pedretti, R. et al. Detection of Circulating Transthyretin Amyloid Aggregates in Plasma: A Novel Biomarker for Transthyretin Amyloidosis. Circulation 149, 1696-1699 (2024). https://doi.org:10.1161/circulationaha.123.067225
9 Pedretti, R. et al. Detection of ATTR aggregates in the plasma of polyneuropathic patients with ATTR-V30M amyloidosis. Amyloid 31, 350-352 (2024). https://doi.org:10.1080/13506129.2024.2404073
10 Zhang, J. et al. A Rare Unusually Aggressive Form of Hereditary Transthyretin Amyloidosis. JACC: Case Reports 30 (2025/10/01). https://doi.org:10.1016/j.jaccas.2025.105309
| Authors : | |
 |
Dr. Rose Pedretti, PhD, Postdoctoral fellow, UT Southwestern Medical Center Dr. Pedretti’s work focuses on developing conformation-specific clinical tools for protein aggregation diseases. Her long-term goal is to bring technologies developed in the Saelices laboratory to the clinic, ultimately improving outcomes for patients with protein aggregation diseases. |
 ; ; |
Pauline Nguyen, PhD Student, UT Southwestern Medical Center Ms. Nguyen’s work focuses on using the conformation-specific clinical tools developed in the Saelices lab to better define and understand the pathogenesis of protein aggregation diseases. Her long-term goal is to develop technologies that can improve patient outcomes. |
 |
Dr. Lorena Saelices Gomez, PhD, Associate Professor, UT Southwestern Medical Center Dr. Saelices’s work focuses on the structural biology of protein misfolding, with a particular emphasis on amyloid systemic diseases. She specializes in developing conformation-specific probes to detect toxic protein aggregates and translating these tools into detection platforms. Her long-term goal is to improve outcomes for patients with underdiagnosed protein aggregation diseases through innovations in biotechnology and translational research. |
| Reviewers : | |
 |
Dr. Mary Cacace, PhD, Postdoctoral Associate, U Pittsburgh. Dr Cacace’s research broadly focuses on therapeutic development at the interface of chemistry and biology. Her graduate training emphasized synthetic chemistry, while her current postdoctoral research centers on ovarian cancer biology, with a particular focus on the role of aldehyde dehydrogenase enzymes in chemoresistance. Dr. Cacace coordinated the review process of this layman summary by Pedretti et al.
|
 |
Alex Roberts, PhD Student, U Pittsburgh Alex Roberts is a second-year graduate student researcher in the PCI-Oncology Graduate Program at the University of Pittsburgh. His research focuses on ovarian cancer biology, with particular emphasis on quiescent cancer cells and their role in promoting chemoresistance.
|
 |
Noor Ahmed, Undergraduate Student, U Pittsburgh Noor is a junior undergraduate at the University of Pittsburgh pursuing a bachelor’s degree in neuroscience, with plans to attend medical school after graduation. Her undergraduate research focuses on ovarian cancer biology. |