Citation Information
Fellner, J Life Sci. Vol. 3, No. 1, March 2021:65-77 https://doi.org/f86n
Crystallography was used to characterize the cesium-binding sites in the sacrificial sulfur transferase LarE from Lactobacillus plantarum. Cs+ binding was unique when compared to a large range of other previously investigated metals. Database searches reveal that Cs+ preferentially binds to protein backbone carbonyl groups, often at crystal contacts and that the use of Cs+ in protein crystallography is an underutilized approach.
Keywords: cesium, caesium, metal binding, crystal contacts, protein crystallography, LarE, ATP pyrophophatase domain, PP-loop, sacrificial sulfur transferase
Introduction
About 30–40% of proteins require one or more metal ions to perform their biological function in cells(1-2) and around a third of the protein structures in the protein data bank (PDB) include one or more metal atoms(3-5). Atomic structures of metal-binding sites often give insights into the function of these proteins. The high selectivity for certain metals at binding sites requires one to screen many elements, and analysis of metal binding to protein crystals is a useful tool for doing this. Here, unique binding sites for cesium (Cs+) in the sacrificial sulfur transferase LarE are presented and differentiating properties of Cs+ binding in protein crystals compared to other metals are discussed.
LarE, together with LarB and LarC(6-9), functions to assemble the nickel-pyridinium-3-thioamide-5-thiocarboxylic acid mononucleotide or nickel-pincer nucleotide (NPN) cofactor(10-11) in around 25% of bacteria(12). While the role of the cofactor in ~15% of bacteria is unknown, the remaining ~10% insert NPN into homologs of LarA(7, 13). LarA homologs use the cofactor to facilitate the racemization of lactate, malate, phenyllactate, α-hydroxyglutarate, or other short-chain aliphatic α-hydroxy acids along with the 2-epimerization of D-gluconate or other sugars (7, 14-15). Of particular interest, two molecules of LarE catalyze sacrificial sulfur insertion reactions that transform their cysteine C176 residues into dehydroalanines while converting pyridinium-3,5-biscarboxylic acid mononucleotide into the NPN precursor pyridinium-3,5-bisthiocarboxylic acid mononucleotide (8, 16-17).
LarE is comprised of a N-terminal PP-loop ATP pyrophosphatase domain that binds and utilizes ATP, a flexible linker containing the catalytic C176 residue, and a C-terminal head domain that facilitates oligomerization(16-18)(Figure 1). The hexameric LarE protein also possesses, separate from the active sites, two metal-binding sites formed by three D231 residues of each trimer, with each carboxylate exhibiting bidentate coordination(16). This tri-Asp site stoichiometrically binds Ca2+, Mn2+, Fe2+/Fe3+, Co2+, Ni2+, Cu2+, Zn2+, and Cd2+, but not Na+, Mg2+, Cl-, K+, Cr3+, Br-, Sr2+ Y3+ , I- , Cs+ or Ba 2+(19). While some metals also showed binding to surface-exposed residues, only Cs+ revealed unique binding sites in the LarE crystal not occupied by any other element. Here these sites are described and placed in the greater context of Cs+ binding in protein crystals.
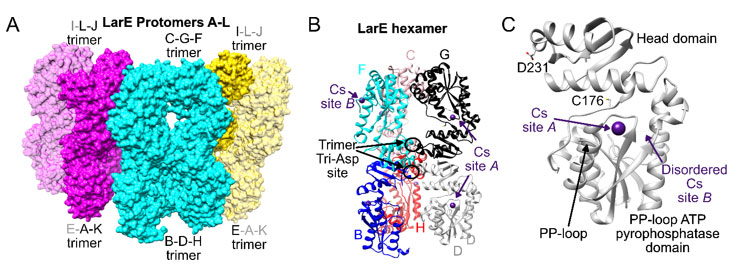
Figure 1 Overall structure of cesium-bound LarE. A) 12 LarE protomers labeled A to L are present in the asymmetric unit and illustrated as surfaces. The central hexamer is shown in cyan and the other two hexamer halves in magenta and yellow. Symmetry related LarE protomer copies completing the hexamer halves are in transparent magenta and yellow. B) Central hexamer in ribbon representation, with every LarE protomer in a different color. Bound Cs+ atoms are depicted in purple, with examples of site A and B indicated. The central D231 tri-Asp trimer site is highlighted. C) Single LarE protomer illustrating the location of Cs+ sites A and B in relationship to the active site C176, PP-loop, and central D231 residue.
Cesium is an alkali metal with the atomic number 55, it is the least electronegative element and has no known biological role. However, it is capable of replacing potassium in the body to some extent because of its chemical similarity. Cesium may be characterized as a sphere of positive charge generally attracting donors with little regard for ligand orientation(20). Key results include the preference of Cs+ for protein backbone carbonyl groups, a clear distinction to other metals and that sites are often found at crystal contacts, indicating a potential for Cs+ binding to improve the quality of protein crystals.
1. Materials and methods
1.1. Expression and purification of LarE
LarE was overexpressed in an Escherichia coli Arctic-Express pBADHisA expression system using construct pGIR076 as previously described(16). Briefly, plasmid transformation and bacterial growth followed the Arctic-Express instruction manual with minor alterations (16). After cell growth in 1 L lysogeny broth at 37 °C to OD600 ~0.4-0.6, the flask was transferred to 13 °C and induced with 0.2% L-arabinose for 24-26 h. The bacterial pellet was harvested and stored at -20 °C. Following cell lysis by sonication, protein purification followed the Strep II-tag affinity chromatography protocol (IBA) with minor adjustments (16). The samples were eluted from Strep-tactin superflow® resin (IBA) in Poly-Prep Chromatography Columns (Bio-Rad) using 100 mM Tris-HCl (pH 7.5) containing 300 mM NaCl, concentrated to ~27 mg/mL, and directly used to set up crystal drops.
1.2. Crystallization of LarE in complex with cesium
For vapor diffusion hanging drop crystallization at 21 °C, 5 µL of ~27 mg/mL LarE in 100 mM Tris-HCl (pH 7.5) containing 300 mM NaCl was mixed with 1 µL 1 M cesium chloride (CsCl, Hampton Additive ScreenTM HR2-428) and 4 µL of reservoir solution. The reservoir contained 100 µL of 30% v/v pentaerythritol ethoxylate (15/4 EO/OH), 50 mM Bis-Tris (pH 6.5), and 100 mM ammonium sulfate, similar to reported conditions(16-17, 19). Crystals were frozen directly in liquid nitrogen without additional cryoprotection. Data was collected at the Life Sciences Collaborative Access Team beamline 21-ID-G at the Advanced Photon Source, Argonne National Laboratory. Tables 1 and 2 list information on data collection, processing, structure solution, and refinement. The dataset was processed with xdsapp(21-22), with merging and scaling performed using aimless(23). Phaser molecular replacement (24) was utilized using the wild-type apoprotein model, PDB ID 5UDQ. Model building and refinement were conducted in Coot(25) and Phenix(24). The Cs-bound structure was deposited under PDB ID 6DG3. UCSF Chimera(26) was used to create structure figures.
|
Diffraction source |
APS 21-ID-G |
|
Wavelength (Å) |
0.979 |
|
Temperature (K) |
100 |
|
Detector |
Marmosaic 300 mm CCD |
|
Space group |
C2221 |
|
a, b, c (Å) |
152.37, 154.16, 328.86 |
|
α, β, γ (°) |
90, 90, 90 |
|
Resolution range (Å) |
48.91–2.94 (3.00–2.94) |
|
No. of unique reflections |
81.489 (4.232) |
|
Completeness (%) |
99.30 (95.00) |
|
Redundancy |
6.20 (6.00) |
|
〈 I/σ(I)〉 |
14.800 (3.7) |
|
Rr.i.m. |
0.123 (0.514) |
|
Rp.i.m. |
0.049 |
|
Overall B factor from Wilson plot (Å2) |
48.62 |
Table 1 Data collection and processing of cesium-bound LarE (PDB ID 6DG3). Values for the outer shell are given in parentheses.
|
Resolution range (Å) |
48.91–2.94 (2.98–2.94) |
|
Completeness (%) |
96.9 |
|
σ cutoff |
F > 1.910σ(F) |
|
No. of reflections, working set |
76777 (4503) |
|
No. of reflections, test set |
7771 (199) |
|
Final Rcryst |
0.196 (0.315) |
|
Final Rfree |
0.274 (0.384) |
|
Protein atoms |
21658 |
|
Cs+ atoms |
12 |
|
Phosphate molecules |
12 |
|
Water |
7 |
|
Total |
21737 |
|
R.m.s. deviations |
|
|
Bonds (Å) |
0.008 |
|
Angles (°) |
1.037 |
|
Average B factors (Å2) |
|
|
Protein |
43.8 |
|
Cs+ |
101.4 |
|
Phosphate |
39.7 |
|
Water |
35.0 |
|
Ramachandran plot |
|
|
Most favoured (%) |
96.78 |
|
Allowed (%) |
3.11 |
Table 2 Structure solution and refinement of cesium-bound LarE (PDB ID 6DG3). Values for the outer shell are given in parentheses.
2.3 Metal analysis tools
The worldwide protein data bank(5) and metalPDB(27) were searched for information on Cs-bound protein structures. The checkmymetal (4) webserver and UCSF Chimera metal geometry (26) were used to examine the LarE and other Cs-bound structures. The Cambridge Structural Database program ConQuest(28) was used to search for small molecule structures containing cesium.
2. Results
Co-crystallization of LarE with CsCl changed the space group for the crystal from P4122 with a single hexamer, observed in nearly all our previously deposited structures for this protein(16-17, (19), to C2221 with two hexamers (across symmetry-related protomers) within the asymmetric unit (Figure 1A). Interestingly this Cs-induced space group shift is also observed when ammonium sulfate was exchanged to thiosulfate in the reported crystallization conditions(19). While these datasets could still be processed using P4122, worse scaling and refining statistics indicated that C2221 was the correct solution. Refinement statistics in C2221 still remained poorer compared to other LarE structures, which resulted in a conservative resolution cut-off at 2.94 Å, despite some scaling parameters indicating that the data could be extended to higher resolution. The packing of the LarE protomers is slightly shifted when comparing the hexamers in P4122 to C2221. This shift influences crystal packing and results in an inferior C2221 crystal form in terms of resulting dataset quality, even though the solvent content is basically identical at ~50% for both crystal forms.
Two unique Cs-binding sites in LarE were discovered in the co-crystallized structure, metal binding at these sites were never observed in any other of the over 250 LarE
|
Site AI: |
Asn53-OD1 |
Ser54-O |
Phe57-O |
Asp59-OD1 |
HOHII |
|
3.71±0.18 |
3.14±0.10 |
3.19±0.17 |
3.38±0.22 |
3.58±0.26 |
|
|
Site BIII |
Glu141-O |
Glu141-N |
Ala144-O |
Ser146-OG |
HOHVI |
|
2.96±0.04 |
3.59±0.22 |
3.25±0.09 |
3.87±0.00 |
3.46±0.13 |
Table 3 {C}Distances in Å of cesium coordinations at site A and site B in LarE (PDB ID 6DG3).
IFor site A Cs+ binding is observed in all chains: Cs+ residue 301.A-L except for chain F and J
IIIn 5 of 10 sites A one water molecule was modelled
IIIFor site B Cs+ binding is only observed in chain F and chain J
IVFor one site B two water molecules were modelled and none in the other site B
crystal datasets examined over the years. For binding site A, Cs+ was bound to the N53-D59 loop (Figure 1B). This surface-exposed loop had Cs+ bound to ten of the twelve LarE protomers (chain A to L) in the asymmetric unit, whereas crystal contacts apparently blocked Cs+ binding to the remaining two copies (no binding in chains F and J). Site A is located between the β2 strand and the α2 helix of the PP-loop ATP pyrophosphatase domain 18 in close proximity to the ATP-binding PP-loop located between β1 and α1 (Figure 1B and C). The active site C176 residue is located even closer on the connecting flexible loop between the PP-loop ATP pyrophosphatase domain and the head domain. The nearby E61 on α2 points towards C176 suggesting a possible role in LarE activity. However, an E61A variant 16 showed unchanged activity, prompting us to believe that this region of the protein only serves a general structural role. The site A Cs+ atom is coordinated by the side chain oxygen atoms of N53 and D59 (Table 3), as well as the backbone oxygen atoms of S54 and F57 (Figure 2A). An additional water molecule is also coordinated to the Cs+ atom. Coordinating atoms refer to all potentially metal-ligating atom within 4.0 Å distance as analyzed by the UCSF chimera 26 metal geometry tool. This tool also allows to predict the most likely geometries ranked by RMSD when all sites in addition to the coordinating atoms would be occupied. However, it has to be stated that the predicted fully hydrated cesium site may also never be observed in protein structures and the possibility remains that Cs+ atoms are able to bind to proteins without full hydration, not following a typical metal geometry. The most likely predicted geometry for this site differs slightly between chains, suggesting a full coordination of 8 or 12, with the most likely geometry being bisdisphenoid (dodecahedral) or anticuboctahedron (triangular orthobicupola). Cs+ binding at site A does not result in any significant changes in this region or overall when compared to unbound LarE structures.
The second Cs-binding location, site B (Figure 1B), involving the E141-S146 loop, was occupied in only 2 out of 12 protomers, (only in chain F and J). As these protomers are the two that did not contain site A, crystal contacts appear to have facilitated site B binding, while preventing site A binding. At site B, Cs+ is bound by the side chain oxygen atom of S146 (Table 3), the backbone oxygen atoms of E141 and A144, and the backbone nitrogen atom of E141, as well as additional water molecules (Figure 2B). The most likely predicted geometry is anticuboctahedron. This loop is located on a section between residue 124 to 154, a region of high variability among members of the PP-loop ATP pyrophosphatase domain(18). This variable region does not appear to be critical for the overall fold of the domain, however it is crucial for activity; i.e., it interacts with the substrate and/or the bound ATP to facilitate the specific reaction of each member enzyme. In the case of LarE, this region includes R133, a residue important for ATP binding and processing(16). This region is flexible and sections fold very differently among LarE protomers, ranging from completely disordered (Figure 3A), to partially disordered and folded into a helix (Figure 3B and 3C), to a
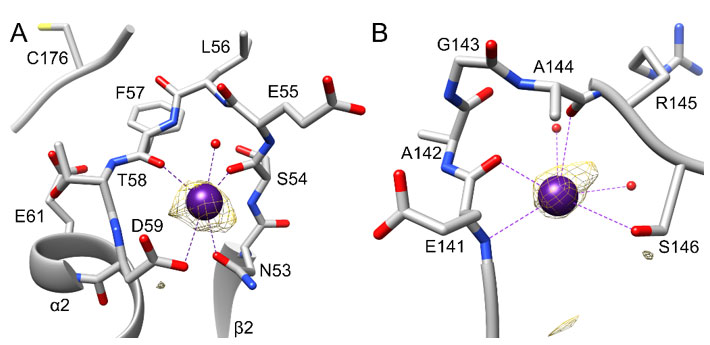
Figure 2 {C}Cesium-binding sites in LarE. The Cs+ atoms and coordination are shown in purple, and bound water atoms are in red. Protein carbon atoms are illustrated in grey, nitrogen atoms in blue, oxygen atoms in red and the sulfur atom in yellow. The anomalous signal map in each region is shown in yellow at 3 σ. A) Cs+ binding site A at the N53-D59 loop. The nearby E61, pointing towards the active site C176 is also is illustrated. B) Cs+ binding site B at the E141-S146 loop.
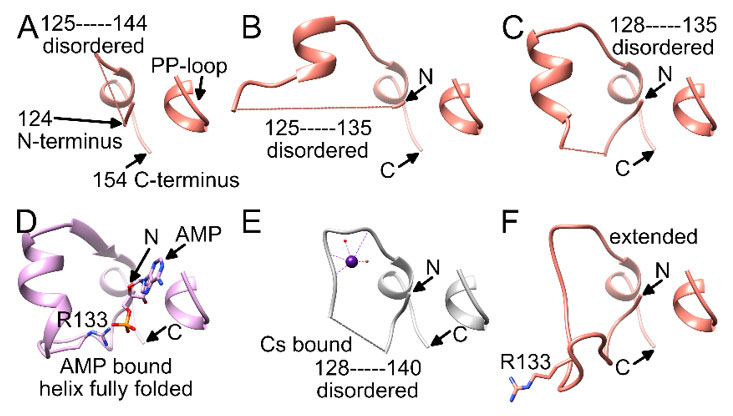
Figure 3 {C}Comparison of the residue 124-154 region in different LarE protomers. A-C) Partially disordered region in different protomers from the LarE apoprotein structure (PDB ID 5UNM). D) Fully folded region with AMP bound to the PP-loop and R133 pairing with AMP (PDB ID 5UDT). E) Cs+ bound at site B with a partially-disordered region (PDB ID 6DG3). F) Fully-folded region in an extended state with R133 pointing in the opposite direction (PDB ID 5UNM).
folded helix which enables R133 to pair with the phosphate group of ATP or AMP after pyrophosphate release (Figure 3D). Binding of Cs+ to site B (Figure 3E) retains disorder in this region, but overall resembles a fully folded extended conformation, with R133 pointing in the opposite direction (Figure 3F). Cs+ binding to site B might restrict the flexibility of this region, which potentially could influence activity.
It has to be noted that the reliability of the presented LarE-Cs structure is not as high as for other LarE-ligand structures due to several factors. First, co-crystallization with Cs+ and the shift to space group C2221 resulted in an overall lower resolution at 2.94 Å compared to most other LarE structures around 2.1-2.5 Å(16). Due to the lower resolution several Cs+ atoms may have incomplete hydration as there was not enough electron density evidence to build additional water ligands. In addition, the B-factors of the Cs+ atoms are very high compared to the surrounding residues, resulting in refinement to or close to 100% occupancy, due to the coupling of B-factors to occupancy at this resolution. Different refinement strategies could result in lower occupancies combined with lower B-factors, but either result and approach indicates lower occupancy or disorder of the exact location of the Cs+ atoms. In general, the refinement of the Cs+ atoms in Phenix was difficult, as there are limited parameters for Cs+ available. After several attempts with different parameters, only bond length restrictions at the default value of 3.0 Å resulted in the observed model. Electron density and especially the observed anomalous map peaks (Figure 2A and 2B) give a lot of confidence that the Cs+ atoms are present, especially when compared to a lack of these signals in any other LarE structure. The presented dataset reflects unique Cs+ binding in LarE; however, due to the many shortcomings mentioned, examination of the data must consider possible model inaccuracies. Analysis by the webserver checkmymetal {C}{C}{C}{C}{C}{C}{C}{C}{C}(4) also reveals high B-factors, incomplete coordination symmetry, and deviations from ideal angles. However, the webserver also notes that many analyses by checkmymetal are hard to validate due to a lack of available high-resolution protein structures with bound cesium.
To gain more insights into cesium binding sites in proteins, all Cs-bound structures in the protein databank (rcsb.org)(5) were examined. Of a total of 139 entries, 109 have unique sequences with 92 of these proteins annotated as bacterial, 40 as eukaryotic, and 7 others. Overall, there is no apparent association of Cs-bound structures to a specific type of protein; e.g., the enzyme classifications are also very diverse with 27 lyases, 17 hydrolases, 16 oxidoreductases, 12 transferases, 7 ligases, and 3 isomerases. The experimental procedure of 57 entries lists the use of CsCl, with an additional 10 specifically mentioning soaking the crystal with CsCl. Hits also include 13 entries with cesium sulfate, 9 using Bicine cesium hydroxide, two with unspecified cesium, one including cesium formate, and one cesium cacodylate. This listing demonstrates that co-crystallization with CsCl as an additive yields the highest chances to obtain a Cs-bound crystal, using a concentration range of 10-200 mM, with 100 mM being used most frequently. In certain cases, CsCl is also listed as a main component of a crystallization cocktail at 2 or 3 M.
MetalPDB(27) lists 639 cesium sites in protein structures, with 138 being unique. Overwhelmingly, these sites are worse modelled than what is reported here for LarE, with the majority lacking in protein coordination and hydration. In a quarter of Cs-binding sites, only water molecules are listed as ligands, often with only a single site of hydration. Removing these sites, as well as sites without any coordination leaves 103 which were analyzed to identified shared features.
Cs+ sites are overwhelmingly bound by the main chain carbonyl groups of protein residues and no specific preference to the nature of the residues appears to exist. 11 structures, with 6 unique sequences of dataset resolution of higher than 1.5 Å were examined in greater detail by hand (Table 4). In these structures nearly all Cs+ atoms show at least one coordinating backbone carbonyl group. Many also show monodentate coordination by a side chain oxygen atom, without a clear preference for a specific amino acid. Infrequent bidentate coordination indicates that such cases might be due to model inaccuracies. In rare occasions interaction by nitrogen backbone or side chain atoms is also observed, however as these interactions have coordination distances of more than 3.5 Å, they might be close contacts rather than important coordinating atoms. If nitrogen atoms are involved in Cs+
|
Site |
Ligands |
H2O |
O |
Other |
Predicted |
||||
|
PDB: Name, overall resolution |
# |
av Å |
# |
av Å |
# |
av Å |
Coordination** |
||
|
4NG8: Lysozyme C, 1.09Å29 |
|||||||||
|
201.A |
O5 |
2 |
3.19 |
2 |
3.09 |
1xAsnOD1 |
2.80 |
8 |
|
|
202.A* |
O3 |
1 |
3.55 |
1 |
3.06 |
1xAsnOD1 |
3.28 |
7 |
|
|
5CGQ: Tryptophan synthase, 1.18Å (unpublished) |
|||||||||
|
302.A |
O8 |
5 |
3.19 |
2 |
2.83 |
1xSerOG |
2.83 |
12 |
|
|
404.B |
O7 |
1 |
2.75 |
6 |
3.14 |
9 |
|||
|
405.B |
O9 |
5 |
3.28 |
3 |
2.96 |
1xThrOG1 |
3.01 |
12 |
|
|
1S3C: Arsenate reductase, 1.25Å(36) |
|||||||||
|
301.A* |
O8 |
2 |
3.34 |
6 |
3.22 |
12 |
|||
|
302.A |
O5 N1 |
3 |
3.10 |
1 |
3.12 |
1xGluOE1 |
3.36 |
12 |
|
|
1xN |
3.90 |
|
|||||||
|
303.A* |
O5 N1 |
4 |
3.61 |
1xGluOE2 |
3.25 |
12 |
|||
|
|
|
|
|
|
|
|
1xGlnNE2 |
3.84 |
|
|
1G57: 3,4-dihydroxy-2-butanone-4-phosphate synthase, 1.40Å(37) |
|||||||||
|
503.A |
O7 |
5 |
3.19 |
2 |
3.35 |
14 |
|||
|
504.A* |
O7 N1 |
6 |
3.28 |
1 |
3.02 |
1xArgNE |
3.57 |
12 |
|
|
1AV2: Gramicidin A, 1.40Å(38) |
|
||||||||
|
18.A |
O7 N2 |
3 |
2.95 |
4 |
3.66 |
2xN |
3.68 |
12 |
|
|
18.B |
O7 N1 |
2 |
2.96 |
5 |
3.59 |
1xN |
3.58 |
12 |
|
|
1UZM: Maba reductase, 1.49Å(30) |
|||||||||
|
1246.A* |
O5 |
3 |
3.49 |
2 |
2.91 |
14 |
|||
|
|
1247.A* |
O10 |
6 |
3.29 |
4 |
3.18 |
|
|
12 |
Table 4 {C}{C}{C}{C}{C}{C}{C}{C}{C}{C}{C}{C}{C}Complete list of high-resolution structures (<1.5 Å) containing Cs+ binding sites. Specific sites identified according to their PDB residue number. O refers to backbone oxygen atom; N refers to backbone nitrogen atom; # indicates the amount of coordinating atoms; av Å stands for the average distance in Å between the Cs+ atom and the coordinating atoms. The predicted coordination number for a fully hydrated site is indicated. Cs+ sites with alternate conformations were omitted.
*site at a crystal contact
**Listed are the coordination numbers predicted by UCSF chimera based on the lowest RMSD to an ideal geometry. All sites that are not fully hydrated have multiple predicted possible geometries and coordination numbers ranked by RMSD. However, cesium sites may never be fully hydrated in protein structures and the observed coordination indicated by the number of ligands represents the final coordination.
coordination, they most likely would only be able to do so when deprotonated, as small molecule compounds do not contain a lot of Cs-H and no Cs-HN entries. In most cases, even for high-resolution structures, full hydration is not modelled, indicating that these sites exist with incomplete hydration. Coordination number predictions for fully hydrated Cs+ binding sites range from 7-14 with various forms of geometry, with 12 for an anticubeoctahedron being observed most frequent. Cs-oxygen distances from water molecules, carbonyl backbone groups or protein side chains vary a lot but appear to mostly fall within 3.3±0.5 Å, indicating that refinement with 3 Å distance constraints is a valid approach.
The highest resolution structure at 1.09 Å (PDB ID 4NG8) originates from an investigation into Rb+, Cs+, Mn2+, Co2+ and Yb3+ binding to lysozyme(29){C}{C}{C}{C}{C}{C}{C}. This investigation also showed that monovalent Rb+ and Cs+ preferentially bind to carbonyl groups, whereas the multivalent Mn2+, Co2+ and Yb3+ interact with carboxylic groups. Some sites show alternate positions for a specific Cs+ atom at the same coordination site, which appear to limit the amount of modelled coordinating water molecules. This could be another reason of why many Cs+ binding sites in protein structures, especially when alternate positions are merged together, lack complete hydration.
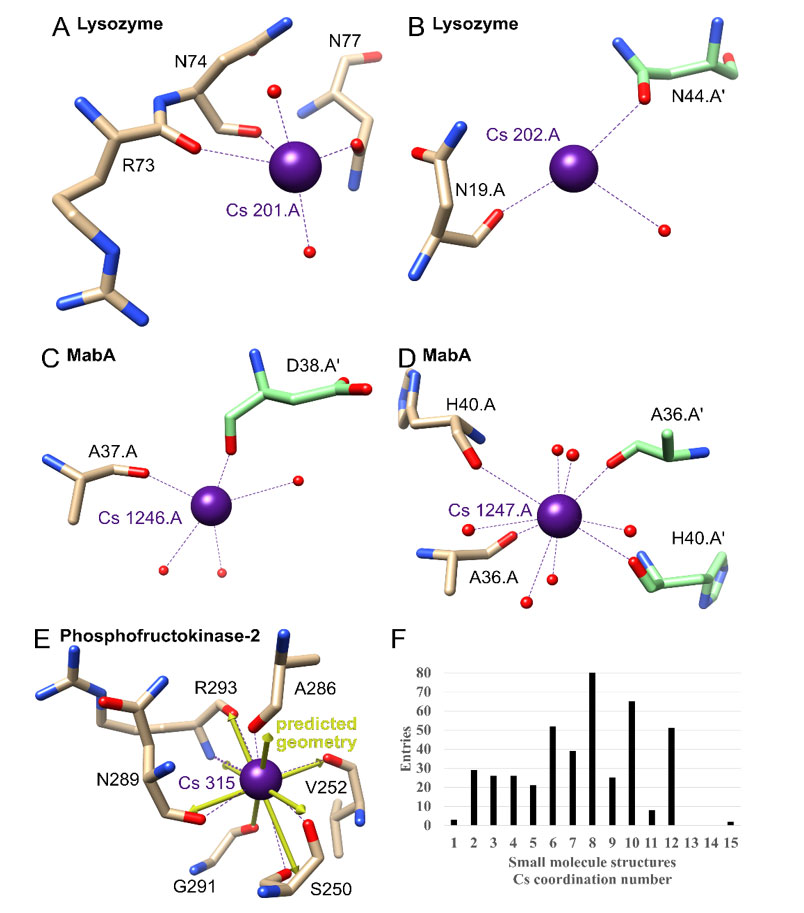
Figure 4 {C}A-E) Examples of cesium-binding sites in other proteins. The Cs+ atoms and coordination are shown in purple, and bound water atoms are in red. Protein carbon atoms are illustrated in brown or for symmetry related copies in green, nitrogen atoms in blue and oxygen atoms in red. A-B) Lysozyme (PDB ID 4NG8). C-D) MabA (1UZM). E) Phosphofructokinase-2 (3UMP) with predicted ideal geometry as yellow arrows. F) Cs+ coordination number in the Cambridge Structural Database for small molecules.
For simplicity Table 4 only lists Cs+ sites for all structures which do not have alternate positions. Lysozyme contains two of these sites, with the first showing coordination by two adjacent backbone carbonyl groups, two water molecules and one oxygen atom from a side chain, in this case from an asparagine residue (Figure 4A). For the possible fully hydrated predicted geometry of bisdisphenoid (8-coordinated), three coordination sites are not occupied. This is an example of the typical coordination and lack of complete hydration of Cs+ sites in protein crystals. The second one is another example, only showing one coordinating backbone oxygen, one water molecule and one side chain oxygen atom from a symmetry related protomer (Figure 4B).
Cs+ binding between protomers within the protein crystal appears to be common, with an example being binding to Mycobacterium tuberculosis MabA (Table 4; 1.49 Å; PDB ID 1UZM)(30). This binding apparently improved the diffraction power of the protein crystals as optimization of the type of chloride salt used in purification and crystallization resulted in a final ranking of Na+<K+<Rb+<Cs+. The positive effect on the crystal quality appears to be based on Cs+ binding at two different sites to backbone carbonyl groups between crystal contacts (Figure 4C and 4D).
Within structures, highly coordinated Cs+ sites are most frequently found in pockets were multiple carbonyl groups face each other. An example for this with a complete hydration is found in Phosphofructokinase-2 from Escherichia coli (1.85 Å; PDB ID 3UMP)(31). Here a Cs+ atom is coordinated by six carbonyl groups, one oxygen side chain atom and one backbone nitrogen atom (although this Cs-N bond is longer than the normally accepted distance for coordination of 4.0 Å at 4.1 Å) to complete the predicted 8-coordinated hexagonal bipyramid geometry (Figure 4E). This is an example where the illustrated predicted geometry vectors in UCSF chimera match the observed ones, something that is more challenging when predicting the full coordination for sites with incomplete hydration, which may not be observed because of lack of data or because a given site may exist with partial hydration (Table 4).
As there is a large range of predicted coordination numbers for cesium in protein structures, small molecule cesium structures were examined next. The Cambridge Structural Database contains 2206 small molecule hits for cesium. 82% of entries contain a Cs-O coordination, followed by 20% Cs-N and 6% Cs-F, with no other element above 4%. The most frequent coordination number (excluding polymeric coordination) is 8, closely followed by 10, 6 and 12 (Figure 4F). The distribution matches what is predicted to be the ideal geometry in the protein structures, only ruling out 14. In general, there does not appear to be a single preferred coordination number which previous analyses of this database in the greater context of metals have also indicated for cesium {C}{C}{C}{C}{C}{C}{C}{C}{C}(32-33).
3. Discussion
Extensive studies have characterized cation interactions with LarE (16, 19). Here two unique Cs-binding sites in LarE are described. It is unlikely that metal interaction with the Cs-binding sites in LarE plays a functional role. Site A is located close to the active site C176 residue and the ATP binding PP-loop, so alkali metals in general could stabilize this loop; however, the loop conformation is indistinguishable when Cs+ is bound and when it is not. The region around site B is very flexible and critical for interaction with ATP. Cs+ binding might restrict this flexibility, which could influence activity.
A main finding is that Cs+ sites in protein crystals occupy unique positions compared to other metals due to their preference for carbonyl backbone groups. LarE is a good example for this, as investigations of over 250 LarE crystal datasets only revealed binding to the carbonyl groups at site A and B when Cs+ was used for co-crystallization. Analyses of high-resolution datasets, containing Cs+ atoms, also clearly show the involvement of carbonyl groups. In addition, Cs+ appears to often bind to exposed backbones at crystal contacts, in many cases between the carbonyl group of the same residue in symmetry related protein copies. In general, it appears that Cs+ sites are mostly modelled as not fully hydrated in protein crystals. They either exist with incomplete hydration or can adopt different geometries with predicted coordination numbers most commonly being 8-12.
Cs-binding sites could be used to solve the phase problem of crystal structures. This possibility is especially useful when Cs+ binds at unique sites compared to other heavy metals, as for the LarE case. Solving the phases in crystal structures via the use of single-wavelength anomalous scattering using Cs+ has been reported(34), but here Cs+ atoms dominate the scattering of these crystals due to the small size of the used protein. The phases for the presented LarE dataset at a wavelength of 0.979 Å could not be solved using the default settings in Phenix autosol and searching for Cs+(35); however, the significant anomalous peak provides confidence that a more focused effort in tuning the wavelength and data collection parameters or the use of a Cu source could lead to solving the phases with Cs+ for these crystals. Co-crystallization with CsCl could be an easy experiment to assist in solving the phases for challenging proteins. Unique Cs-binding sites may also be accessible via metal soaking.
Overall, there are not enough high-resolution structures to clearly identify the most common Cs+ geometry and coordination in protein crystals, as well as any preference for certain side chain oxygen atoms over others. However, the main distinguishing characteristic of Cs+ binding sites is their preference for carbonyl backbone groups and their ability to bind to or even facilitate crystal contacts. This should encourage protein crystallography experiments with Cs+ for many other proteins to identify unique binding sites, similar to the approach taken for LarE.
4. References
|
1. Andreini, C.; Bertini, I.; Rosato, A., Metalloproteomes: a bioinformatic approach. Acc Chem Res 2009, 42, 1471-1479. https://doi.org/10.1021/ar900015x PMid:19697929 |
|
|
2. Andreini, C.; Bertini, I.; Cavallaro, G.; Holliday, G. L.; Thornton, J. M., Metal ions in biological catalysis: from enzyme databases to general principles. J Biol Inorg Chem 2008, 13, 1205-1218. https://doi.org/10.1007/s00775-008-0404-5 PMid:18604568 |
|
|
3. Harding, M. M.; Nowicki, M. W.; Walkinshaw, M. D., Metals in protein structures: a review of their principal features. Crystallogr Rev 2010, 16, 247-302. https://doi.org/10.1080/0889311X.2010.485616 |
|
|
4. Zheng, H.; Chordia, M. D.; Cooper, D. R.; Chruszcz, M.; Muller, P.; Sheldrick, G. M.; Minor, W., Validation of metal-binding sites in macromolecular structures with the CheckMyMetal web server. Nat Protoc 2014, 9, 156-170. https://doi.org/10.1038/nprot.2013.172 PMid:24356774 PMCid:PMC4410975 |
|
|
5. Berman, H.; Henrick, K.; Nakamura, H.; Markley, J. L., The worldwide Protein Data Bank (wwPDB): ensuring a single, uniform archive of PDB data. Nucleic Acids Res 2007, 35, D301-303. https://doi.org/10.1093/nar/gkl971 PMid:17142228 PMCid:PMC1669775 |
|
|
6. Desguin, B.; Goffin, P.; Viaene, E.; Kleerebezem, M.; Martin-Diaconescu, V.; Maroney, M. J.; Declercq, J. P.; Soumillion, P.; Hols, P., Lactate racemase is a nickel-dependent enzyme activated by a widespread maturation system. Nat Commun 2014, 5, 3615. https://doi.org/10.1038/ncomms4615 PMid:24710389 PMCid:PMC4066177 |
|
|
7. Desguin, B.; Zhang, T.; Soumillion, P.; Hols, P.; Hu, J.; Hausinger, R. P., A tethered niacin-derived pincer complex with a nickel-carbon bond in lactate racemase. Science 2015, 349, 66-69. https://doi.org/10.1126/science.aab2272 PMid:26138974 |
|
|
8. Desguin, B.; Soumillion, P.; Hols, P.; Hausinger, R. P., Nickel-pincer cofactor biosynthesis involves LarB-catalyzed pyridinium carboxylation and LarE-dependent sacrificial sulfur insertion. Proc Natl Acad Sci U S A 2016, 113, 5598-5603. https://doi.org/10.1073/pnas.1600486113 PMid:27114550 PMCid:PMC4878509 |
|
|
9. Desguin, B.; Fellner, M.; Riant, O.; Hu, J.; Hausinger, R. P.; Hols, P.; Soumillion, P., Biosynthesis of the nickel-pincer nucleotide cofactor of lactate racemase requires a CTP-dependent cyclometallase. J Biol Chem 2018, 293, 12303-12317. https://doi.org/10.1074/jbc.RA118.003741 PMid:29887527 PMCid:PMC6093250 |
|
|
10. Hausinger, R. P.; Desguin, B.; Fellner, M.; Rankin, J. A.; Hu, J., Nickel-pincer nucleotide cofactor. Curr Opin Chem Biol 2018, 47, 18-23. https://doi.org/10.1016/j.cbpa.2018.06.019 PMid:30015232 |
|
|
11. Nevarez, J.; Turmo, A.; Hu, J.; Hausinger, R. P., Biological pincer complexes. ChemCatChem 2020, 12, 4242-4254. https://doi.org/10.1002/cctc.202000575 PMid:33072225 |
|
|
12. Desguin, B., Lactate Racemization and Beyond. J Bacteriol Parasitol 2018, 9, 2. https://doi.org/10.4172/2155-9597.1000335 |
|
|
13. Fellner, M.; Rankin, J. A.; Hu, J.; Hausinger, R. P., Lactate Racemase. Encyclopedia of Inorganic and Bioinorganic Chemistry 2017, 1-8. https://doi.org/10.1002/9781119951438.eibc2508 |
|
|
14. Desguin, B.; Urdiain-Arraiza, J.; Da Costa, M.; Fellner, M.; Hu, J.; Hausinger, R. P.; Desmet, T.; Hols, P.; Soumillion, P., Uncovering a superfamily of nickel-dependent hydroxyacid racemases and epimerases. Sci. Rep. 2020, 10, 18123. https://doi.org/10.1038/s41598-020-74802-6 PMid:33093595 PMCid:PMC7583248 |
|
|
15. Rankin, J. A.; Mauban, R. C.; Fellner, M.; Desguin, B.; McCracken, J.; Hu, J.; Varganov, S. A.; Hausinger, R. P., Lactate Racemase Nickel-Pincer Cofactor Operates by a Proton-Coupled Hydride Transfer Mechanism. Biochemistry 2018, 57, 3244-3251. https://doi.org/10.1021/acs.biochem.8b00100 PMid:29489337 |
|
|
16. Fellner, M.; Desguin, B.; Hausinger, R. P.; Hu, J., Structural insights into the catalytic mechanism of a sacrificial sulfur insertase of the N-type ATP pyrophosphatase family, LarE. Proc Natl Acad Sci U S A 2017, 114, 9074-9079. https://doi.org/10.1073/pnas.1704967114 PMid:28784764 PMCid:PMC5576804 |
|
|
17. Fellner, M.; Rankin, J. A.; Desguin, B.; Hu, J.; Hausinger, R. P., Analysis of the Active Site Cysteine Residue of the Sacrificial Sulfur Insertase LarE from Lactobacillus plantarum. Biochemistry 2018, 57, 5513-5523. https://doi.org/10.1021/acs.biochem.8b00601 PMid:30157639 |
|
|
18. Fellner, M.; Hausinger, R. P.; Hu, J., A structural perspective on the PP-loop ATP pyrophosphatase family. Crit Rev Biochem Mol Biol 2018, 53, 607-622. DOI: https://doi.org/10.1080/10409238.2018.1516728 PMid:30280944 |
|
|
19. Fellner, M.; Huizenga, K. G.; Hausinger, R. P.; Hu, J., Crystallographic characterization of a tri-Asp metal-binding site at the three-fold symmetry axis of LarE. Sci. Rep. 2020, 10, 5830. https://doi.org/10.1038/s41598-020-62847-6 PMid:32242052 PMCid:PMC7118094 |
|
|
20. Bryan, J. C.; Kavallieratos, K.; Sachleben, R. A., Unusual Ligand Coordination for Cesium. Inorganic Chemistry 2000, 39, 1568-1572. https://doi.org/10.1021/ic991203v PMid:12526466 |
|
|
21. Sparta, K. M.; Krug, M.; Heinemann, U.; Mueller, U.; Weiss, M. S., Xdsapp2.0. J. Appl. Crystallogr. 2016, 49, 1085-1092. https://doi.org/10.1107/S1600576716004416 |
|
|
22. Kabsch, W., Xds. Acta Crystallogr D Biol Crystallogr 2010, 66, 125-132. https://doi.org/10.1107/S0907444909047337PMid:20124692 PMCid:PMC2815665 |
|
|
23. Evans, P. R.; Murshudov, G. N., How good are my data and what is the resolution? Acta Crystallogr D 2013, 69, 1204-1214. DOI: 10.1107/S0907444913000061. |
|
|
24. Adams, P. D.; Afonine, P. V.; Bunkoczi, G.; Chen, V. B.; Davis, I. W.; Echols, N.; Headd, J. J.; Hung, L. W.; Kapral, G. J.; Grosse-Kunstleve, R. W.; McCoy, A. J.; Moriarty, N. W.; Oeffner, R.; Read, R. J.; Richardson, D. C.; Richardson, J. S.; Terwilliger, T. C.; Zwart, P. H., PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 2010, 66, 213-221. https://doi.org/10.1107/S0907444909052925 PMid:20124702 PMCid:PMC2815670 |
|
|
25. Emsley, P.; Lohkamp, B.; Scott, W. G.; Cowtan, K., Features and development of Coot. Acta Crystallogr D Biol Crystallogr 2010, 66, 486-501. https://doi.org/10.1107/S0907444910007493PMid:20383002 PMCid:PMC2852313 |
|
|
26. Pettersen, E. F.; Goddard, T. D.; Huang, C. C.; Couch, G. S.; Greenblatt, D. M.; Meng, E. C.; Ferrin, T. E., UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 2004, 25, 1605-1612. https://doi.org/10.1002/jcc.20084 PMid:15264254 |
|
|
27. Putignano, V.; Rosato, A.; Banci, L.; Andreini, C., MetalPDB in 2018: a database of metal sites in biological macromolecular structures. Nucleic Acids Res 2018, 46, D459-D464. https://doi.org/10.1093/nar/gkx989 PMid:29077942 PMCid:PMC5753354 |
|
|
28. Bruno, I. J.; Cole, J. C.; Edgington, P. R.; Kessler, M.; Macrae, C. F.; McCabe, P.; Pearson, J.; Taylor, R., New software for searching the Cambridge Structural Database and visualizing crystal structures. Acta Crystallographica Section B 2002, 58, 389-397. DOI: https://doi.org/10.1107/S0108768102003324 PMid:12037360 |
|
|
29. Benas, P.; Auzeil, N.; Legrand, L.; Brachet, F.; Regazzetti, A.; Ries-Kautt, M., Weak protein-cationic co-ion interactions addressed by X-ray crystallography and mass spectrometry. Acta Crystallographica Section D 2014, 70, 2217-2231. https://doi.org/10.1107/S1399004714011304 PMid:25084340 |
|
|
30. Cohen-Gonsaud, M.; Ducasse, S.; Hoh, F.; Zerbib, D.; Labesse, G.; Quemard, A. k., Crystal Structure of MabA from Mycobacterium tuberculosis, a Reductase involved in Long-chain Fatty Acid Biosynthesis. J Mol Biol 2002, 320, 249-261. https://doi.org/10.1016/S0022-2836(02)00463-1 |
|
|
31. Baez, M.; Cabrera, R.; Pereira, Humberto M.; Blanco, A.; Villalobos, P.; Ramírez-Sarmiento, César A.; Caniuguir, A.; Guixé, V.; Garratt, Richard C.; Babul, J., A Ribokinase Family Conserved Monovalent Cation Binding Site Enhances the MgATP-induced Inhibition in E. coli Phosphofructokinase-2. Biophysical Journal 2013, 105, 185-193. https://doi.org/10.1016/j.bpj.2013.05.028 PMid:23823238 PMCid:PMC3699746 |
|
|
32. Dudev, M.; Wang, J.; Dudev, T.; Lim, C., Factors governing the metal coordination number in metal complexes from Cambridge Structural Database analyses. J Phys Chem B 2006, 110, 1889-1895. https://doi.org/10.1021/jp054975n PMid:16471760 |
|
|
33. Kuppuraj, G.; Dudev, M.; Lim, C., Factors Governing Metal−Ligand Distances and Coordination Geometries of Metal Complexes. The Journal of Physical Chemistry B 2009, 113, 2952-2960. https://doi.org/10.1021/jp807972e PMid:19708219 |
|
|
34. Wallace, B.; Hendrickson, W.; Ravikumar, K., The use of single-wavelength anomalous scattering to solve the crystal structure of a gramicidin A/caesium chloride complex. Acta Crystallogr., Sect. B: Struct. Sci. 1990, 46, 440-446. https://doi.org/10.1107/S0108768190001161 PMid:1696485 |
|
|
35. Terwilliger, T. C.; Adams, P. D.; Read, R. J.; McCoy, A. J.; Moriarty, N. W.; Grosse-Kunstleve, R. W.; Afonine, P. V.; Zwart, P. H.; Hung, L. W., Decision-making in structure solution using Bayesian estimates of map quality: the PHENIX AutoSol wizard. Acta Crystallogr D Biol Crystallogr 2009, 65, 582-601. https://doi.org/10.1107/S0907444909012098 PMid:19465773 PMCid:PMC2685735 |
|
|
36. DeMel, S.; Shi, J.; Martin, P.; Rosen, B. P.; Edwards, B. F. P., Arginine 60 in the ArsC arsenate reductase of E. coli plasmid R773 determines the chemical nature of the bound As(III) product. Protein Science 2004, 13, 2330-2340. https://doi.org/10.1110/ps.04787204 PMid:15295115 PMCid:PMC2280019 |
|
|
37. Liao, D.-I.; Calabrese, J. C.; Wawrzak, Z.; Viitanen, P. V.; Jordan, D. B., Crystal Structure of 3,4-Dihydroxy-2-Butanone 4-Phosphate Synthase of Riboflavin Biosynthesis. Structure 2001, 9, 11-18. https://doi.org/10.1016/S0969-2126(00)00550-5 |
|
|
38. Burkhart, B. M.; Li, N.; Langs, D. A.; Pangborn, W. A.; Duax, W. L., The conducting form of gramicidin A is a right-handed double-stranded double helix. Proceedings of the National Academy of Sciences 1998, 95, 12950-12955. https://doi.org/10.1073/pnas.95.22.12950 PMid:9789021 PMCid:PMC23667 |
References
|
1. Andreini, C.; Bertini, I.; Rosato, A., Metalloproteomes: a bioinformatic approach. Acc Chem Res 2009, 42, 1471-1479. https://doi.org/10.1021/ar900015x PMid:19697929 | |
|
2. Andreini, C.; Bertini, I.; Cavallaro, G.; Holliday, G. L.; Thornton, J. M., Metal ions in biological catalysis: from enzyme databases to general principles. J Biol Inorg Chem 2008, 13, 1205-1218. https://doi.org/10.1007/s00775-008-0404-5 PMid:18604568 | |
|
3. Harding, M. M.; Nowicki, M. W.; Walkinshaw, M. D., Metals in protein structures: a review of their principal features. Crystallogr Rev 2010, 16, 247-302. https://doi.org/10.1080/0889311X.2010.485616 | |
|
4. Zheng, H.; Chordia, M. D.; Cooper, D. R.; Chruszcz, M.; Muller, P.; Sheldrick, G. M.; Minor, W., Validation of metal-binding sites in macromolecular structures with the CheckMyMetal web server. Nat Protoc 2014, 9, 156-170. https://doi.org/10.1038/nprot.2013.172 PMid:24356774 PMCid:PMC4410975 | |
|
5. Berman, H.; Henrick, K.; Nakamura, H.; Markley, J. L., The worldwide Protein Data Bank (wwPDB): ensuring a single, uniform archive of PDB data. Nucleic Acids Res 2007, 35, D301-303. https://doi.org/10.1093/nar/gkl971 PMid:17142228 PMCid:PMC1669775 | |
|
6. Desguin, B.; Goffin, P.; Viaene, E.; Kleerebezem, M.; Martin-Diaconescu, V.; Maroney, M. J.; Declercq, J. P.; Soumillion, P.; Hols, P., Lactate racemase is a nickel-dependent enzyme activated by a widespread maturation system. Nat Commun 2014, 5, 3615. https://doi.org/10.1038/ncomms4615 PMid:24710389 PMCid:PMC4066177 | |
|
7. Desguin, B.; Zhang, T.; Soumillion, P.; Hols, P.; Hu, J.; Hausinger, R. P., A tethered niacin-derived pincer complex with a nickel-carbon bond in lactate racemase. Science 2015, 349, 66-69. https://doi.org/10.1126/science.aab2272 PMid:26138974 | |
|
8. Desguin, B.; Soumillion, P.; Hols, P.; Hausinger, R. P., Nickel-pincer cofactor biosynthesis involves LarB-catalyzed pyridinium carboxylation and LarE-dependent sacrificial sulfur insertion. Proc Natl Acad Sci U S A 2016, 113, 5598-5603. https://doi.org/10.1073/pnas.1600486113 PMid:27114550 PMCid:PMC4878509 | |
|
9. Desguin, B.; Fellner, M.; Riant, O.; Hu, J.; Hausinger, R. P.; Hols, P.; Soumillion, P., Biosynthesis of the nickel-pincer nucleotide cofactor of lactate racemase requires a CTP-dependent cyclometallase. J Biol Chem 2018, 293, 12303-12317. https://doi.org/10.1074/jbc.RA118.003741 PMid:29887527 PMCid:PMC6093250 | |
|
10. Hausinger, R. P.; Desguin, B.; Fellner, M.; Rankin, J. A.; Hu, J., Nickel-pincer nucleotide cofactor. Curr Opin Chem Biol 2018, 47, 18-23. https://doi.org/10.1016/j.cbpa.2018.06.019 PMid:30015232 | |
|
11. Nevarez, J.; Turmo, A.; Hu, J.; Hausinger, R. P., Biological pincer complexes. ChemCatChem 2020, 12, 4242-4254. https://doi.org/10.1002/cctc.202000575 PMid:33072225 | |
|
12. Desguin, B., Lactate Racemization and Beyond. J Bacteriol Parasitol 2018, 9, 2. https://doi.org/10.4172/2155-9597.1000335 | |
|
13. Fellner, M.; Rankin, J. A.; Hu, J.; Hausinger, R. P., Lactate Racemase. Encyclopedia of Inorganic and Bioinorganic Chemistry 2017, 1-8. https://doi.org/10.1002/9781119951438.eibc2508 | |
|
14. Desguin, B.; Urdiain-Arraiza, J.; Da Costa, M.; Fellner, M.; Hu, J.; Hausinger, R. P.; Desmet, T.; Hols, P.; Soumillion, P., Uncovering a superfamily of nickel-dependent hydroxyacid racemases and epimerases. Sci. Rep. 2020, 10, 18123. https://doi.org/10.1038/s41598-020-74802-6 PMid:33093595 PMCid:PMC7583248 | |
|
15. Rankin, J. A.; Mauban, R. C.; Fellner, M.; Desguin, B.; McCracken, J.; Hu, J.; Varganov, S. A.; Hausinger, R. P., Lactate Racemase Nickel-Pincer Cofactor Operates by a Proton-Coupled Hydride Transfer Mechanism. Biochemistry 2018, 57, 3244-3251. https://doi.org/10.1021/acs.biochem.8b00100 PMid:29489337 | |
|
16. Fellner, M.; Desguin, B.; Hausinger, R. P.; Hu, J., Structural insights into the catalytic mechanism of a sacrificial sulfur insertase of the N-type ATP pyrophosphatase family, LarE. Proc Natl Acad Sci U S A 2017, 114, 9074-9079. https://doi.org/10.1073/pnas.1704967114 PMid:28784764 PMCid:PMC5576804 | |
|
17. Fellner, M.; Rankin, J. A.; Desguin, B.; Hu, J.; Hausinger, R. P., Analysis of the Active Site Cysteine Residue of the Sacrificial Sulfur Insertase LarE from Lactobacillus plantarum. Biochemistry 2018, 57, 5513-5523. https://doi.org/10.1021/acs.biochem.8b00601 PMid:30157639 | |
|
18. Fellner, M.; Hausinger, R. P.; Hu, J., A structural perspective on the PP-loop ATP pyrophosphatase family. Crit Rev Biochem Mol Biol 2018, 53, 607-622. DOI: https://doi.org/10.1080/10409238.2018.1516728 PMid:30280944 | |
|
19. Fellner, M.; Huizenga, K. G.; Hausinger, R. P.; Hu, J., Crystallographic characterization of a tri-Asp metal-binding site at the three-fold symmetry axis of LarE. Sci. Rep. 2020, 10, 5830. https://doi.org/10.1038/s41598-020-62847-6 PMid:32242052 PMCid:PMC7118094 | |
|
20. Bryan, J. C.; Kavallieratos, K.; Sachleben, R. A., Unusual Ligand Coordination for Cesium. Inorganic Chemistry 2000, 39, 1568-1572. https://doi.org/10.1021/ic991203v PMid:12526466 | |
|
21. Sparta, K. M.; Krug, M.; Heinemann, U.; Mueller, U.; Weiss, M. S., Xdsapp2.0. J. Appl. Crystallogr. 2016, 49, 1085-1092. https://doi.org/10.1107/S1600576716004416 | |
|
22. Kabsch, W., Xds. Acta Crystallogr D Biol Crystallogr 2010, 66, 125-132. https://doi.org/10.1107/S0907444909047337PMid:20124692 PMCid:PMC2815665 | |
|
23. Evans, P. R.; Murshudov, G. N., How good are my data and what is the resolution? Acta Crystallogr D 2013, 69, 1204-1214. DOI: 10.1107/S0907444913000061. |
|
|
24. Adams, P. D.; Afonine, P. V.; Bunkoczi, G.; Chen, V. B.; Davis, I. W.; Echols, N.; Headd, J. J.; Hung, L. W.; Kapral, G. J.; Grosse-Kunstleve, R. W.; McCoy, A. J.; Moriarty, N. W.; Oeffner, R.; Read, R. J.; Richardson, D. C.; Richardson, J. S.; Terwilliger, T. C.; Zwart, P. H., PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 2010, 66, 213-221. https://doi.org/10.1107/S0907444909052925 PMid:20124702 PMCid:PMC2815670 |
|
|
25. Emsley, P.; Lohkamp, B.; Scott, W. G.; Cowtan, K., Features and development of Coot. Acta Crystallogr D Biol Crystallogr 2010, 66, 486-501. https://doi.org/10.1107/S0907444910007493PMid:20383002 PMCid:PMC2852313 |
|
|
26. Pettersen, E. F.; Goddard, T. D.; Huang, C. C.; Couch, G. S.; Greenblatt, D. M.; Meng, E. C.; Ferrin, T. E., UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 2004, 25, 1605-1612. https://doi.org/10.1002/jcc.20084 PMid:15264254 |
|
|
27. Putignano, V.; Rosato, A.; Banci, L.; Andreini, C., MetalPDB in 2018: a database of metal sites in biological macromolecular structures. Nucleic Acids Res 2018, 46, D459-D464. https://doi.org/10.1093/nar/gkx989 PMid:29077942 PMCid:PMC5753354 |
|
|
28. Bruno, I. J.; Cole, J. C.; Edgington, P. R.; Kessler, M.; Macrae, C. F.; McCabe, P.; Pearson, J.; Taylor, R., New software for searching the Cambridge Structural Database and visualizing crystal structures. Acta Crystallographica Section B 2002, 58, 389-397. DOI: https://doi.org/10.1107/S0108768102003324 PMid:12037360 |
|
|
29. Benas, P.; Auzeil, N.; Legrand, L.; Brachet, F.; Regazzetti, A.; Ries-Kautt, M., Weak protein-cationic co-ion interactions addressed by X-ray crystallography and mass spectrometry. Acta Crystallographica Section D 2014, 70, 2217-2231. https://doi.org/10.1107/S1399004714011304 PMid:25084340 |
|
|
30. Cohen-Gonsaud, M.; Ducasse, S.; Hoh, F.; Zerbib, D.; Labesse, G.; Quemard, A. k., Crystal Structure of MabA from Mycobacterium tuberculosis, a Reductase involved in Long-chain Fatty Acid Biosynthesis. J Mol Biol 2002, 320, 249-261. https://doi.org/10.1016/S0022-2836(02)00463-1 |
|
|
31. Baez, M.; Cabrera, R.; Pereira, Humberto M.; Blanco, A.; Villalobos, P.; Ramírez-Sarmiento, César A.; Caniuguir, A.; Guixé, V.; Garratt, Richard C.; Babul, J., A Ribokinase Family Conserved Monovalent Cation Binding Site Enhances the MgATP-induced Inhibition in E. coli Phosphofructokinase-2. Biophysical Journal 2013, 105, 185-193. https://doi.org/10.1016/j.bpj.2013.05.028 PMid:23823238 PMCid:PMC3699746 |
|
|
32. Dudev, M.; Wang, J.; Dudev, T.; Lim, C., Factors governing the metal coordination number in metal complexes from Cambridge Structural Database analyses. J Phys Chem B 2006, 110, 1889-1895. https://doi.org/10.1021/jp054975n PMid:16471760 |
|
|
33. Kuppuraj, G.; Dudev, M.; Lim, C., Factors Governing Metal−Ligand Distances and Coordination Geometries of Metal Complexes. The Journal of Physical Chemistry B 2009, 113, 2952-2960. https://doi.org/10.1021/jp807972e PMid:19708219 |
|
|
34. Wallace, B.; Hendrickson, W.; Ravikumar, K., The use of single-wavelength anomalous scattering to solve the crystal structure of a gramicidin A/caesium chloride complex. Acta Crystallogr., Sect. B: Struct. Sci. 1990, 46, 440-446. https://doi.org/10.1107/S0108768190001161 PMid:1696485 |
|
|
35. Terwilliger, T. C.; Adams, P. D.; Read, R. J.; McCoy, A. J.; Moriarty, N. W.; Grosse-Kunstleve, R. W.; Afonine, P. V.; Zwart, P. H.; Hung, L. W., Decision-making in structure solution using Bayesian estimates of map quality: the PHENIX AutoSol wizard. Acta Crystallogr D Biol Crystallogr 2009, 65, 582-601. https://doi.org/10.1107/S0907444909012098 PMid:19465773 PMCid:PMC2685735 |
|
|
36. DeMel, S.; Shi, J.; Martin, P.; Rosen, B. P.; Edwards, B. F. P., Arginine 60 in the ArsC arsenate reductase of E. coli plasmid R773 determines the chemical nature of the bound As(III) product. Protein Science 2004, 13, 2330-2340. https://doi.org/10.1110/ps.04787204 PMid:15295115 PMCid:PMC2280019 |
|
|
37. Liao, D.-I.; Calabrese, J. C.; Wawrzak, Z.; Viitanen, P. V.; Jordan, D. B., Crystal Structure of 3,4-Dihydroxy-2-Butanone 4-Phosphate Synthase of Riboflavin Biosynthesis. Structure 2001, 9, 11-18. https://doi.org/10.1016/S0969-2126(00)00550-5 |
|
|
38. Burkhart, B. M.; Li, N.; Langs, D. A.; Pangborn, W. A.; Duax, W. L., The conducting form of gramicidin A is a right-handed double-stranded double helix. Proceedings of the National Academy of Sciences 1998, 95, 12950-12955. https://doi.org/10.1073/pnas.95.22.12950 PMid:9789021 PMCid:PMC23667 |