Abstract Human immunodeficiency virus (HIV) preferentially infects T-lymphocytes by integrating into host DNA and forming a latent transcriptionally silent provirus. As previously shown, HIV-1 alters migration modes of T-lymphocytes by co-regulating viral gene expression with human C-X-C chemokine receptor-4 (CXCR4). Here, we show that motility of infected T-lymphocytes is cell size dependent. In cell migration assays, migrating cells are consistently larger than non-migrating cells. This effect is drug-treatment independent. The cell size dependent motility observed in a previously generated Jurkat latency model correlates with the motility of primary human CD4+ T-cells containing a modified HIV-1 full-length construct JLatd2GFP. In addition, large migrating T-cells, latently infected with HIV, show a slightly decreased rate of reactivation from latency. these results demonstrate that HIV reactivation is cell migration-dependent, where host cell size acts as a catalyst for altered migration velocity. We believe that host cell size controlled migration uncovers an additional mechanism of cellular controlled viral fate determination important for virus dissemination and reactivation from latency. This observation may provide more insights into viral-host interactions regulating cell migration and reactivation from latency and helps in the design and implementation of novel therapeutic strategies.
Keywords: Cell migration, cell size, HIV-infected T-lymphocytes, reactivation, HIV latency
T-lymphocyte migration is essential for T-cell responses. The expression of CXCR4, on activated CD4+ T-cells, promotes recruitment to the area where chemokine ligand 12 (CXCL12) is expressed. Binding of CXCL12 to human C-X-C motif chemokine receptor-4 (CXCR4) mediates intracellular signaling, mobilization of migrating T-cells into the periphery and cell survival 1 CXCL12 abundant regions enriched with uninfected T-cells are prone to viral infection and propagation. A human immunodeficiency virus (HIV)-infection of CD4+ T-cells causes either the activation of the HIV long terminal repeat (LTR) promoter and the expression of viral proteins, like the trans-activator of transcription (Tat) protein, or leads to the development of latent provirus reservoirs. A viral controlled effect on cell migration has been recently reported 2. This study shows that reactivation of CD4+ T-cells latently infected with HIV decouples a positive correlation of LTR expression and cell migration resulting in decreased cell motility. A genetically coupled CXCR4 promoter and protein interaction at the cell surface between the viral protein Tat was reported as the cause of reactivation-migration coupling in latently infected T-cells 2. Changes in motility of reactivated latent cells pose challenges for the elimination of the latent HIV reservoir due to the accumulation of reactivated cells in target-rich cell niches high in CXCL12 ligand. The observation of cell-size dependent reactivation of T-cells latently infected with HIV, caused by increased gene expression burst from the HIV LTR promoter, may present a novel target for controlling or managing latent reservoirs of infected cells 3. However, investigations of the effect of cell size on migration and its relation to HIV latency have not been performed. Implementing a comprehensive cell size comparison of non-migrating and migrating cells, we discover a cell size-dependent migration effect in T-cells latently infected with HIV. We show that migrating cells of a reactivated cell population are larger while the average cell population size is smaller. This effect is consistent across drug-treated T-cells. Moreover, the %ON of reactivated smaller non-migrating cells is higher than for migrating cells. Finally, the cell size of migrating cells can be differently controlled using drug treatments. Along with our previous findings 2,3, these results reveal cell size as an important component for controlling migration and reactivation towards potentially managing HIV pathogenesis.
Materials and Methods
Cell lines and transfection
JLat full-length clone 15.4 was obtained through the NIH AIDS Reagent Program from E. Verdin. The isoclone was selected from a previously generated library (Jordan et al. 2003). The production of JLatd2GFP lentiviral particles was performed using 5 x 105 HEK293 cells and FuGENE6 transfection reagent (Promega). The transfection was carried out according to manufacturer’s instruction. All transduced cells were harvested 24h after media change and the viral supernatant was centrifugated at 500 x g to remove all remaining cells. Concentration of lentiviral supernatant was archived using a lentivirus concentrator reagent (Takara Bio Inc., CA, USA) and a centrifugation step at 1,500 x g for 45min at 4°C. The lentivirus was concentrated in ice-cold DMEM (Thermo Scientific) supplemented with 10%FBS and 1% penicillin/streptomycin using a ratio of 1:80 of the original volume.
For freshly activated primary CD4+ T-cells, activation beads in cell suspension were no removed and cells infection was performed using a construct consisting of full-length HIV with a destabilized GFP replacing the nef reading frame and a deletion of env (JLatd2GFP). For infection, 60µl of concentrated lentiviral suspension was added to 1 x 106 activated CD4+ T-cells and topped with 40 µl lukewarm RPMI 1640 medium supplemented with 10% FBS and 1% penicillin/streptomycin. Cells and virus were centrifugated at 1,200 x g for 2h at room temperature. Beads were removed after spinoculation using a magnet. Cells were resuspended in RPMI 1640 medium containing 10% FBS, 1% penicillin/streptomycin and 30 U ml-1 interleukin 2.
Cell culture and growth conditions
Jurkats cells were cultured in RPMI 1640 with L-glutamine and 25mM HERPES (Thermo Scientific). The media was supplemented with 10% fetal bovine serum (FBS) and 1% penicillin and streptomycin (Corning Cellgro). All cells were cultured at 37°C with 5% CO2. CD4+ T-cells were isolated from fresh human whole blood (Innovative Research, MI, USA) by collecting buffy coat followed by Ficoll-Hypague density gradient centrifugation. A negative selection was carried out to isolate CD4+ T-cells using RossetteSep Human CD4+ T-Cell Enrichment Kit (Stemcell Technologies). Activation of primary CD4+ T-cells was conducted on the same day of isolation using 1x 106 CD4+ T-cells per ml, 30 u ml-1 human interleukin 2 (Miltenyi Biotec Inc., CA, USA) and 1x 106 Dynabeads Human T-Activator CD3/CD28 (Thermo Fisher Scientific). Cells were activated for 3d.
Sorting of CD4* T-cells
GFP+ and GFP– infected primary CD4+ T-cells were sorted using a primary anti-human CD4+ monoclonal (SIM.4) antibody (NIH AIDS Reagent Program, MD, USA) and a secondary goat anti-mouse IgG-PE antibody (Santa Cruz Biotechnology, TX, USA). Cells were stained for 30 min at 4°C and 20 min at 4°C with the primary and secondary antibody, respectively. All sorting experiments were performed on a BD FACS Aria II.
Drug Treatments
TNF, at a final concentration of 10 ng ml-1, was used for stimulation of the latent Jurkat isoclone 15.4. Romidepsin and Panobinostat, two anticancer drugs, were used at a final concentration of 5 nM and 15 nM, respectively. Iono and JQ1 were used at a final concentration of 1 µM, Prostratin (Pro) at 3 µM, AZA at 5 µM, E2 and Tam at 10 µM and valproic acid at 1 mM. Histone deacetylase inhibitors Trichostatin A (TSA) and SAHA were applied at a final concentration of 400 nM and 2.5 µM, respectively. Phorbol myristate acetate (PMA) was used at a final concentration of 200 ng ml-1. Primary CD4+ T-cells were treated and stimulated with TNF, SAHA, Iono, and a combination of Iono and PMA using the same concentrations listed above. All chemicals were obtained from Cyman Chemicals, except for TSA (Sigma-Aldrich) and TNF (R&D Systems).
Migration assay
Migration assays were performed as previously described in Bohn-Wippert et al. 2017. Briefly, reactivation and cell size investigation experiments were performed using 96-well transwell plates with a pore size of 5 µm (Corning Inc., Product 3388). 1 x 106 cells per ml for the JLat isoclone 15.4 and 0.08 x 106 cells per ml for the stimulated primary CD4+ T-cells were transferred to a 24-well plate (JLat) or 96-V bottom plate (CD4+ T-cells) and treated with a single chemical or a combination of two chemicals for 48h in 37°C and 5% CO2. Cells were pelleted after drug treatment using a centrifugation step of 500 x g and suspended in migration medium RPMI 1640 with L-glutamine and 25 mM HEPES supplemented with 0.5% BSA. 0.2 x 106 cells per 100 µl were seeded for a 96-well migration assay using JLat 15.4., and 0.05 x 106 cells per ml for primary CD4+ T-cells. Human CXCL-12 (or SDF-1, R&D Systems) were diluted to a final concentration of 25 ng ml-1 and added to 0.15 ml migration media of the lower chamber of a transwell plate. Cells were allowed to migrate for 3h at 37°C and 5% CO2. Cell size of non-migrating cells was measured before migration started using the automated cell counter MOXI Z (ORFLO). Cell size of migrating cells, the content of the lower bottom well, was measured after the migration assay has been stopped using the same instrument listed above. The percentage of fluorescent cells among the migrating subpopulation was analyzed at the flow cytometer and for calculating the percentage of migrating GFP-expressing cells using the following formula was conducted:
Total number of GFP+ migrated cells = (% GFP+ of unsorted infected CD4+ T-cells in the bottom of the transwell) x (total number of migrating cells in the bottom transwell).
To validate the viability of treated T-cells at the time of seeding into transwell plates, a propidium iodide staining was performed from each drug concentration. Except for sorted and infected primary CD4+ T-cell treated with TNF, all migration experiments were carried in duplicate, triplicate or quadruplicate on separate days and the average values were plotted.
Flow cytometry analysis
The investigation of the fluorescent GFP intensity of migrating and non-migrating cells was performed as described in Bohn-Wippert et al. 2017. In short, reactivation of migrating and non-migrating cells were measured after 48h post treatment. JLat isoclone 15.4 and infected primary CD4+ T-cells were gated using side scatter versus GFP intensity to differentiate between GFP-negative and GFP-positive cells, compared with conservative gating determined by both NaÏve Jurkats (ATCC clone E6-1) and untreated JLat isoclone 15.4 samples as well as uninfected stimulated and untreated CD4+ T-cells. Ten thousand cells in the live gate were collected using FSC vs. SSC.
Results
Migrating HIV-infected T-lymphocytes are larger than non-migrating cells
To research how cell size has an effect on reactivation and migration, migration assays using chemotactic gradients for the CXCR4-CXCL12 migration axis were performed 2. Isoclone 15.4 of a previously generated Jurkat latency model (JLAT) 4, a Jurkat cell line containing full-length HIV with a deletion of env and a green fluorescent protein (GFP) replacing the nef reading frame, was used (Fig. 1A). Cells were treated with diverse drugs, like tumor-necrosis factor alpha (TNF) or histone deacetylase inhibitor (HDACi) Suberoylanilide hydroxamic acid (SAHA) for 48h. The rate of migration, cell sizes of non-migrating and migrating cells and mean fluorescence of GFP were measured and results were compared to untreated samples (Fig. 1A). Measuring the average mean population size pre-migration, the cell size was smaller with increased rate of reactivation from HIV latency, defined as %ON, and their motility was reduced. In contrast, migrating cells were consistently larger than non-migrating cells and reactivation was decreased (Fig. 1B). Rate of reactivation (%ON) revealed to be drug-dependent. These findings indicate that the more cells reactivate the Macintosh HD:Users:Kathl:Arbeit:AG Dar:Publications:Cell size vs migration:Figures:Figure 1_02062020.pngsmaller their non-migrating cells are.
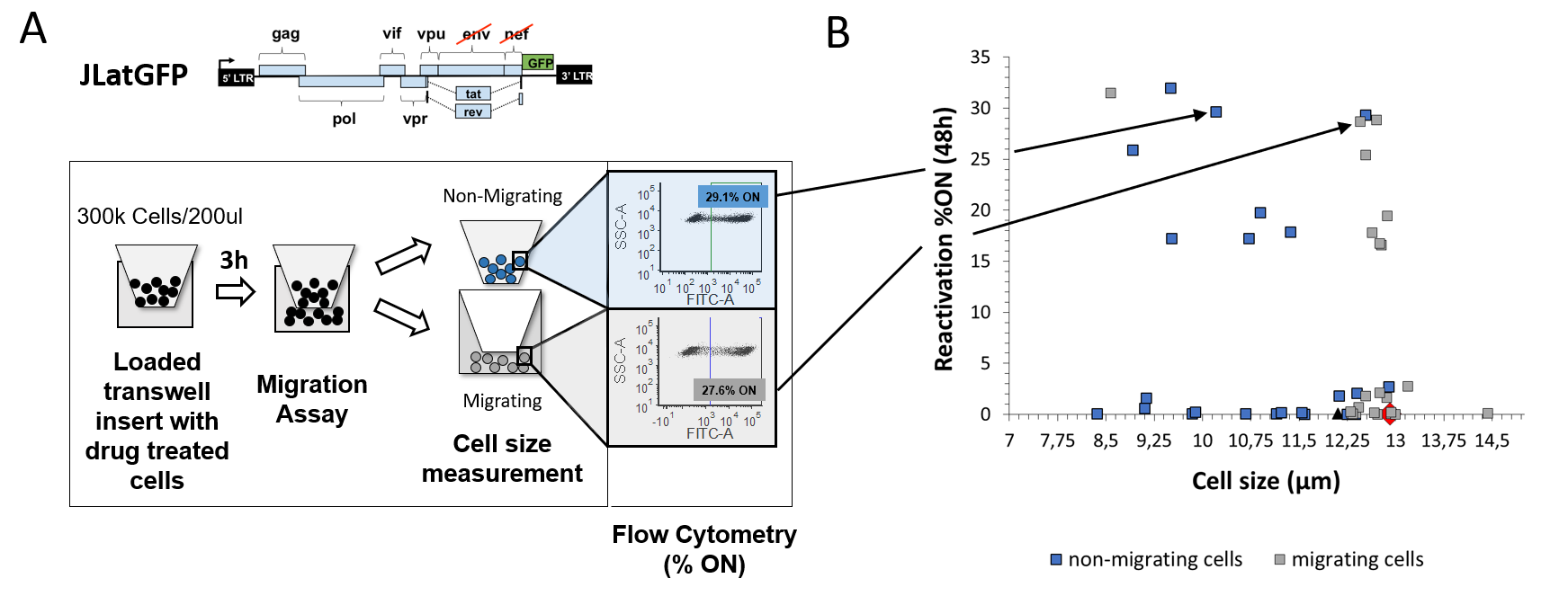
Fig. 1: Migration of T-cells latently infected with HIV is cell size dependent.
(A) Schematic of performance of migration assay and measurement of cell size and flow cytometry. To test migration of latent T-cells infected with HIV, an isoclone 15.4 containing the full-length HIV-1 with deletion of env and GFP replacing the nef reading frame (JLatGFP) was used and treated with diverse drugs for 48h. Afterwards, cells were seeded into a 96-well transwell chamber at a concentration of 300k cells/ 200µl and cell size of seeded cells was measured. 3h after migration, cell size and mean fluorescence of GFP (%ON of reactivation) for non-migrating (blue dots) and migrating (grey dots) cells were analyzed using an automated cell counter and flow cytometry, respectively.
(B) Cell size and reactivation rate (%ON) measurements of the latent T-cell isoclone 15.4 revealed an increase in cell size for migrating cells (grey dots) when compared with their non-migrating counterpart (blue squares). Rate of reactivation is drug and migration dependent. An example of cell size and reactivation differences for the treatment TNF+JQ1 is represented in more detail (black arrows). Untreated samples were color-coded as a black triangle (non-migrating) and a red diamond (migrating). All measurements were performed in duplicate, triplicate or quadruplicate on separate days and the average values and standard errors were plotted.
Drug-treatment alters cell size-dependent migration
To confirm that cell size is capable of altering migration of latent T-cells, exogenous treatment with reactivation drug cocktails were used to observe migration behavior of cells. Cells were treated for 48h with common modulators of HIV transcription as described in Bohn-Wippert et al. 2 and cell size of the cell population was measured before and after migration assays were conducted. Although CXCR4 internalization mechanism at the cell surface after Suberoylanilide hydroxamic acid (SAHA) treatment has been reported 5, and up- and down-regulation effects of CXCR4 expression for drugs like JQ1, Tamoxifen (Tam), 17β-Estradiol (E2) and 5-Aza-2-deoxycytidine (AZA) were described 6, migrating cells were consistently larger than non-migrating cells (Fig. 2). Additionally, changes of cell size before and after migration are drug treatment dependent. Interestingly, a cell size increase after treatment with Cytarabine could be confirmed 9, while the difference of cell size before and after migration was still present. This result reveals a dominant effect of cell size-dependent migration, irrespective of the drug being used, its effect on cell size, and the concentration of CXCR4 at the cell surface.
Robust cell size-dependent migration of HIV-infected primary human CD4+ T-cells
To prove that migrating cells are also larger in primary human cells, migration assays and cell size measurements were performed using GFP-
positive sorted and drug treated HIV-1 infected primary CD4+ T-cells (Fig. 3A). To exclude the possibility of cell toxicity after drug treatment, we used commonly used drug concentrations published in Bohn-Wippert et al. (2). Cells were treated with synergistic activators of latency such as TNF, SAHA and Phorbol 12-myristate 13-acetate (PMA) in combination with Ionomycin (Iono) and results were compared with untreated samples. Irrespective of drug treatment, reactivated CD4+ T-cells showed robust increases in cell size after migration while average cell population size was smaller (Fig. 3B). This result is consistent with drug treatments on Jurkat latency model isoclone 15.4 (Fig. 2).
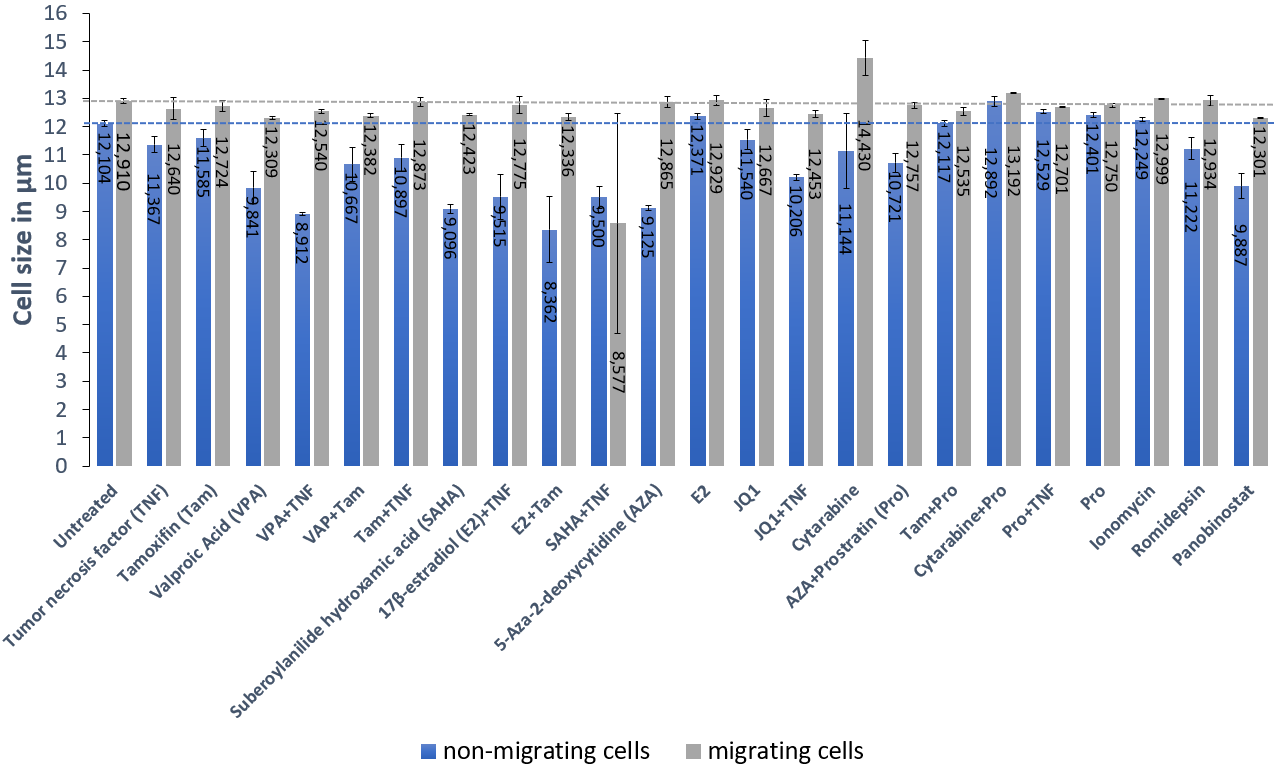
Fig. 2: Migrating cells are larger than non-migrating cells irrespective of drug treatment.
Measurements of cell size for non-migrating (blue bars) and migrating cells (grey bars) of the latent T-cell isoclone 15.4 after 48h of drug treatment reveals an increase of cell size for migrating cells when compared to non-migrating cells. For cell size comparison, the dashed lines represent the size of the untreated JLat isoclone 15.4 for non-migrating (blue) and migrating cells (grey). Value of left most bars. All measurements were performed in duplicate, triplicate or quadruplicate on separate days and the average values and standard errors were plotted
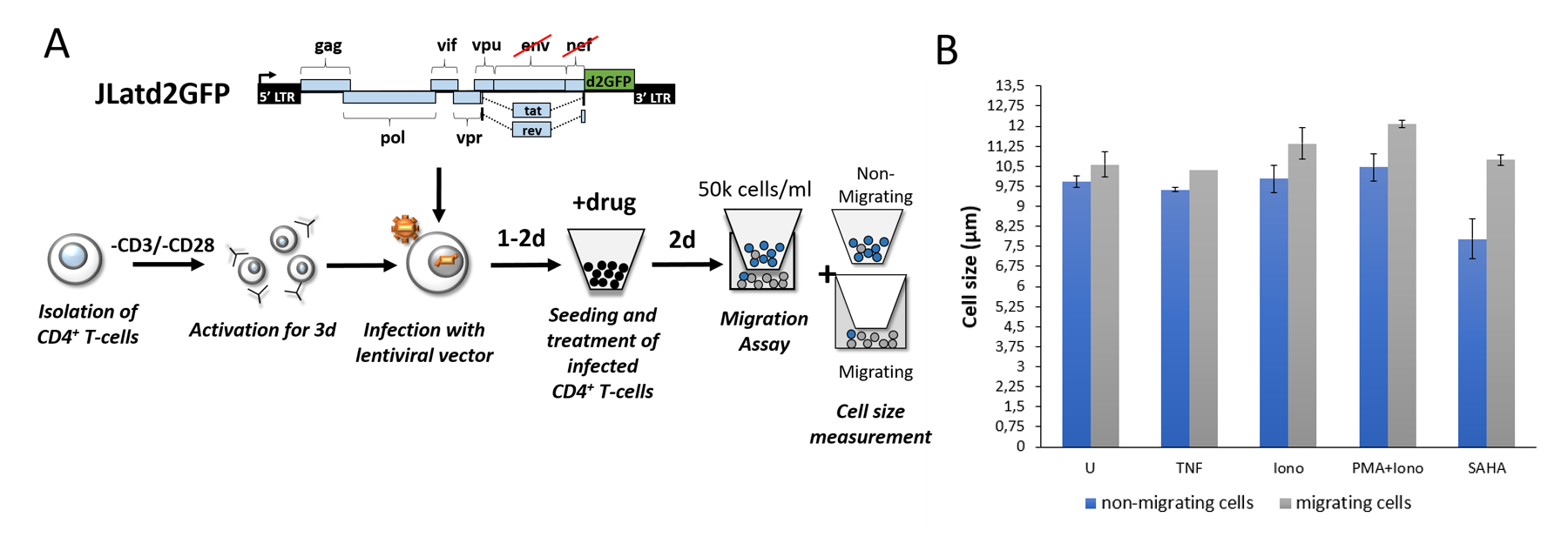
Fig. 3: Cell size of migrating and reactivated HIV-infected primary CD4+ T-cells is increased.
(A) Diagram of infection of primary CD4+ T-cells and performance of migration assay and measurement of cell size and flow cytometry. Primary CD4+ T-cells are isolated from fresh human whole blood and stimulated with anti-CD3/anti-CD28 antibodies on the same day. The cells were infected with concentrated lentivirus containing the full-length HIV-1 genome (JLatd2GFP) 3 days after stimulation. Afterwards, CD4+ T-cells are sorted for GFP-positive cells 1 day after infection. Sorted CD4+ T-cells were drug treated for 48h and seeded into a 96-well transwell chamber at a concentration of 50k cells/ml while cell size was measured for seeded cells. 3h after migration, cell size and %ON of reactivation of migrated cells was measured. (B) GFP-positive sorted primary CD4+ T-cells, infected with the construct JLatd2GFP, were treated with different drugs for 48h and cell size was measured. An increased cell size for migrating cells (grey bars) was observed when compared to non-migrating cells (blue bars). All measurements were performed in duplicate, except of the drug TNF for migrating cells (grey bar), and standard error bars are plotted.
Discussion
We have investigated how changes in cell size regulate migration of latent T-cells. An inverse relationship between viral reactivation and cell size for migrating cells is revealed, where larger cells show higher motility with decreased rates of reactivation. We discover that the rate of migrating cells defines the cell size of non-migrating cells. In brief, the higher the rates of migration and reactivation, the smaller their non-migrating cells are. This suggests a host-induced cell size adjustment of the mean cell size of a cell population. Cell size based impacts on viral expression and decision-making have been recently shown 3. Viral gene expression burst was demonstrated to be cell size dependent. In this study, larger T-cells latently infected with HIV exclusively reactivated from latency. This result presents a cell state capable of passive control of stochastic viral decision making and cell fate. Our result of cell size controlled migration uncovers an additional mechanism of cellular controlled viral fate determination. It has not been previously shown whether cell size is involved in migration velocity of HIV infected T-cells. Here, we show a reactivation dependent viral behavior associated with decelerated cell migration. This observation is of importance for understanding viral spread and virus host-cell interactions. Controlled cell motility in response to external conditions in the environment has been reported for cells, like B-lymphocytes, which adapt their motility behavior to their environment 10. Changing cell size due to an infection state of a cell to control cell motility represents a new mechanism for cell adaptation in response to adverse intracellular changes.
Adaptation to environmental changes does not only occur at the cellular level. Viruses are also capable to adapt to their hosts by taking over cell expression for their own benefit. A recent study suggests a decoupling of migration in full-length models of proviral latency by co-expression of the viral protein Tat with CXCR4 receptor causing decelerated migration of infected cells 2. This suggests that the virus has evolved to synchronize viral expression with host cell CXCR4 up-regulation, to control or decrease CXCR4-mediated migration individually within each infected cell, potentially for a fitness increase (2,11). In this study a consistent difference of cell size for non-migrating and migrating cells and their inverse relationship of reactivation may be under the influence of gene expression of viral proteins within the infected cell. All experiments used full-length latency models expressing the viral protein Tat. Tat transactivation is heterogeneous in clonal populations (12, 13) and larger cells express higher Tat levels than smaller cells when a virus switches from a latent state to an actively transcribing state 3. A negative impact of viral infection and viral gene expression on cell motility has been shown recently. Verolett et al. report that HIV-1 enhances virus spreading in confined environments by reprogrammed and reduced T-cell motility 14. Moreover, cell motility is also controlled by cell adhesion. Previous studies have reported that leukocytes and fibroblasts migrate on different substrates through adhesion 15, and alteration of cell size attributes differences of cell adhesion which may influence migration velocity
Our findings on host-cell size dependent deceleration of migration and reactivation of migrating HIV-infected T-cells may provide insights into viral-host interactions, relationships, HIV pathogenesis and the implementation of novel therapeutic strategies.
Acknowledgements
K. B-W. and R. D. D. acknowledge support by NIH NIAID (AI120746) and NSF CAREER (1943740). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. JLat full-length clone 15.4 was obtained through the NIH AIDS Reagent Program from E. Verdin. We thank L. Weinberger for the kind gift of plasmid JLatd2GFP.
References
1.Doring Y; Pawig L; Weber C; Noels H. The CXCL12/CXCR4 chemokine ligand/receptor axis in cardiovascular disease. Front Physiol. 2014;5:212. https://doi.org/10.3389/fphys.2014.00212PMid:24966838 PMCid:PMC4052746
2. Bohn-Wippert K; Tevonian EN; Megaridis MR; Dar RD. Similarity in viral and host promoters couples viral reactivation with host cell migration. Nat Commun. 2017;8:15006.https://doi.org/10.1038/ncomms15006PMid:28462923 PMCid:PMC5418578
Bohn-Wippert K; Tevonian EN; Lu Y; Huang MY; Megaridis MR; Dar RD. Cell Size-Based Decision-Making of a Viral Gene Circuit. Cell Rep. 2018;25(13):3844-57 e5. https://doi.org/10.1016/j.celrep.2018.12.009 PMid:30590053
4. Jordan A; Bisgrove D; Verdin E. HIV reproducibly establishes a latent infection after acute infection of T cells in vitro. EMBO J. 2003;22(8):1868-77. https://doi.org/10.1093/emboj/cdg188 PMid:12682019 PMCid:PMC154479
5. Clift IC; Bamidele AO; Rodriguez-Ramirez C; Kremer KN; Hedin KE. beta-Arrestin1 and distinct CXCR4 structures are required for stromal derived factor-1 to downregulate CXCR4 cell-surface levels in neuroblastoma. Mol Pharmacol. 2014;85(4):542-52. https://doi.org/10.1124/mol.113.089714 PMid:24452472 PMCid:PMC4170118
6. Banerjee C; Archin N; Michaels D; Belkina AC; Denis GV; Bradner J, et al. BET bromodomain inhibition as a novel strategy for reactivation of HIV-1. J Leukoc Biol. 2012;92(6):1147-54. https://doi.org/10.1189/jlb.0312165 PMid:22802445 PMCid:PMC3501896
7. Kubarek L; Jagodzinski PP. Epigenetic up-regulation of CXCR4 and CXCL12 expression by 17 beta-estradiol and tamoxifen is associated with formation of DNA methyltransferase 3B4 splice variant in Ishikawa endometrial adenocarcinoma cells. FEBS Lett. 2007;581(7):1441-8. https://doi.org/10.1016/j.febslet.2007.02.070 PMid:17362937
8. Yamamoto Y; Pahwa R; Pahwa S. S-nitrosoglutathione modulates CXCR4 and ICOS expression. Cell Mol Biol Lett. 2006;11(1):30-6. https://doi.org/10.2478/s11658-006-0003-9 PMid:16847746 PMCid:PMC6276017
9. Miettinen TP; Bjorklund M. Mevalonate Pathway Regulates Cell Size Homeostasis and Proteostasis through Autophagy. Cell Rep. 2015;13(11):2610-20. https://doi.org/10.1016/j.celrep.2015.11.045 PMid:26686643 PMCid:PMC4709259
10. Rey-Barroso J; Calovi DS; Combe M; German Y; Moreau M; Canivet A, et al. Switching between individual and collective motility in B lymphocytes is controlled by cell-matrix adhesion and inter-cellular interactions. Sci Rep. 2018;8(1):5800. https://doi.org/10.1038/s41598-018-24222-4 PMid:29643414 PMCid:PMC5895587
11. Murooka TT; Deruaz M; Marangoni F; Vrbanac VD; Seung E; von Andrian UH, et al. HIV-infected T cells are migratory vehicles for viral dissemination. Nature. 2012;490(7419):283-7. https://doi.org/10.1038/nature11398 PMid:22854780 PMCid:PMC3470742
12. Weinberger LS; Burnett JC; Toettcher JE; Arkin AP; Schaffer DV. Stochastic gene expression in a lentiviral positive-feedback loop: HIV-1 Tat fluctuations drive phenotypic diversity. Cell. 2005;122(2):169-82. https://doi.org/10.1016/j.cell.2005.06.006 PMid:16051143
13. Weinberger LS; Dar RD; Simpson ML. Transient-mediated fate determination in a transcriptional circuit of HIV. Nat Genet. 2008;40(4):466-70. https://doi.org/10.1038/ng.116 PMid:18344999
14. Verollet C; Le Cabec V; Maridonneau-Parini I. HIV-1 Infection of T Lymphocytes and Macrophages Affects Their Migration via Nef. Front Immunol. 2015;6:514. https://doi.org/10.3389/fimmu.2015.00514 PMid:26500651 PMCid:PMC4594015
15. Trepat X; Chen Z; Jacobson K. Cell migration. Compr Physiol. 2012;2(4):2369-92. https://doi.org/10.1002/cphy.c110012 PMid:23720251 PMCid:PMC4457291
16. Deman JJ; Vakaet LC; Bruyneel EA. Cell size and mutual cell adhesion. II. Evidence for a relation between cell size, long-range electrostatic repulsion and intercellular adhesiveness during density-regulated growth in suspension. J Membr Biol. 1976;26(2-3):205-15. https://doi.org/10.1007/BF01868874 PMid:1263252